|
|
IV.A.4. (XIV.F.) |
Relative enthalpies of isomers - Comparison of 298.15K enthalpies (kJ mol-1)
Isomers of C2H4
| index | Species | CAS number | Name | Relative experimental enthalpy (kJ mol-1) | sketch |
|---|---|---|---|---|---|
| a | CH3CH | 4218502 | methylmethylene | 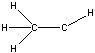 |
|
| b | C2H4 | 74851 | Ethylene | 0.0 | 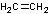 |
| semi-empirical | PM3 | 92.0 a |
|---|---|---|
| MNDOd | 0.0 b |
|
| composite | G1 | 302.8 a 0.0 b |
| G2MP2 | 300.4 a 0.0 b |
|
| G2 | 318.3 a 0.0 b |
|
| G3 | 291.1 a 0.0 b |
|
| G3B3 | 166.1 a 0.0 b |
|
| G3MP2 | 302.5 a 0.0 b |
|
| G4 | 178.4 a 0.0 b |
|
| CBS-Q | 162.5 a 0.0 b |
|
| molecular mechanics | MM3 | 0.0 b |
| STO-3G | 3-21G | 3-21G* | 6-31G | 6-31G* | 6-31G** | 6-31+G** | 6-311G* | 6-311G** | 6-31G(2df,p) | 6-311+G(3df,2p) | 6-311+G(3df,2pd) | TZVP | cc-pVDZ | cc-pVTZ | cc-pVQZ | aug-cc-pVDZ | aug-cc-pVTZ | aug-cc-pVQZ | cc-pV(T+d)Z | cc-pCVDZ | cc-pCVTZ | Sadlej_pVTZ | daug-cc-pVDZ | daug-cc-pVTZ | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hartree fock | HF | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
170.2 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
177.8 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
180.5 a 0.0 b |
| density functional | LSDA | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||||||||||||||
| BLYP | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
293.0 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||
| B1B95 | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||
| B3LYP | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
290.2 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|
| B3LYPultrafine | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| B3PW91 | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||
| mPW1PW91 | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||
| M06-2X | 0.0 b |
0.0 b |
284.9 a 0.0 b |
0.0 b |
281.0 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| PBEPBE | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|
| PBEPBEultrafine | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| PBE1PBE | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
268.1 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| HSEh1PBE | 0.0 b |
271.7 a 0.0 b |
0.0 b |
0.0 b |
267.4 a 0.0 b |
0.0 b |
268.2 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
273.4 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| TPSSh | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
264.5 a 0.0 b |
0.0 b |
265.3 a 0.0 b |
0.0 b |
0.0 b |
266.2 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
270.0 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| wB97X-D | 0.0 b |
0.0 b |
283.8 a 0.0 b |
0.0 b |
280.0 a 0.0 b |
0.0 b |
280.7 a 0.0 b |
0.0 b |
284.4 a 0.0 b |
0.0 b |
0.0 b |
285.2 a 0.0 b |
267.8 a 0.0 b |
286.5 a 0.0 b |
0.0 b |
0.0 b |
285.9 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||||
| B97D3 | 0.0 b |
284.3 a 0.0 b |
0.0 b |
0.0 b |
279.7 a 0.0 b |
0.0 b |
281.3 a 0.0 b |
0.0 b |
283.9 a 0.0 b |
0.0 b |
286.0 a 0.0 b |
285.4 a 0.0 b |
0.0 b |
285.9 a 0.0 b |
0.0 b |
0.0 b |
284.8 a 0.0 b |
0.0 b |
0.0 b |
|||||||
| STO-3G | 3-21G | 3-21G* | 6-31G | 6-31G* | 6-31G** | 6-31+G** | 6-311G* | 6-311G** | 6-31G(2df,p) | 6-311+G(3df,2p) | 6-311+G(3df,2pd) | TZVP | cc-pVDZ | cc-pVTZ | cc-pVQZ | aug-cc-pVDZ | aug-cc-pVTZ | aug-cc-pVQZ | cc-pV(T+d)Z | cc-pCVDZ | cc-pCVTZ | Sadlej_pVTZ | daug-cc-pVDZ | daug-cc-pVTZ | ||
| Moller Plesset perturbation | MP2 | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
285.2 a 0.0 b |
0.0 b |
0.0 b |
288.0 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
292.6 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
| MP2=FULL | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||
| ROMP2 | 0.0 b |
|||||||||||||||||||||||||
| MP3 | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| MP3=FULL | 275.9 a 0.0 b |
278.9 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||||||||||||||||||
| MP4 | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| MP4=FULL | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| B2PLYP | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
286.2 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
295.3 a 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| B2PLYP=FULL | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||||
| Configuration interaction | CID | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| CISD | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||||||
| STO-3G | 3-21G | 3-21G* | 6-31G | 6-31G* | 6-31G** | 6-31+G** | 6-311G* | 6-311G** | 6-31G(2df,p) | 6-311+G(3df,2p) | 6-311+G(3df,2pd) | TZVP | cc-pVDZ | cc-pVTZ | cc-pVQZ | aug-cc-pVDZ | aug-cc-pVTZ | aug-cc-pVQZ | cc-pV(T+d)Z | cc-pCVDZ | cc-pCVTZ | Sadlej_pVTZ | daug-cc-pVDZ | daug-cc-pVTZ | ||
| Quadratic configuration interaction | QCISD | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||
| QCISD(T) | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| QCISD(T)=FULL | 0.0 b |
0.0 b |
||||||||||||||||||||||||
| QCISD(TQ) | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||||||||||
| Coupled Cluster | CCD | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| CCSD | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| CCSD=FULL | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| CCSD(T) | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||
| CCSD(T)=FULL | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||
| STO-3G | 3-21G | 3-21G* | 6-31G | 6-31G* | 6-31G** | 6-31+G** | 6-311G* | 6-311G** | 6-31G(2df,p) | 6-311+G(3df,2p) | 6-311+G(3df,2pd) | TZVP | cc-pVDZ | cc-pVTZ | cc-pVQZ | aug-cc-pVDZ | aug-cc-pVTZ | aug-cc-pVQZ | cc-pV(T+d)Z | cc-pCVDZ | cc-pCVTZ | Sadlej_pVTZ | daug-cc-pVDZ | daug-cc-pVTZ |
| CEP-31G | CEP-31G* | CEP-121G | CEP-121G* | LANL2DZ | SDD | cc-pVTZ-PP | aug-cc-pVTZ-PP | Def2TZVPP | ||
|---|---|---|---|---|---|---|---|---|---|---|
| hartree fock | HF | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
180.5 a 0.0 b |
||
| density functional | LSDA | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| BLYP | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| B1B95 | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| B3LYP | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
291.0 a 0.0 b |
|||
| B3LYPultrafine | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| B3PW91 | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| mPW1PW91 | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| M06-2X | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| PBEPBE | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
287.5 a 0.0 b |
|||
| PBEPBEultrafine | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| PBE1PBE | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| HSEh1PBE | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| TPSSh | 0.0 b |
|||||||||
| wB97X-D | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| B97D3 | 0.0 b |
|||||||||
| Moller Plesset perturbation | MP2 | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
301.0 a 0.0 b |
||
| MP2=FULL | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| MP3 | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| MP4 | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| MP4=FULL | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| B2PLYP | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| B2PLYP=FULL | 0.0 b |
|||||||||
| Configuration interaction | CID | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| CISD | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| Quadratic configuration interaction | QCISD | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| QCISD(T) | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| QCISD(TQ) | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| Coupled Cluster | CCD | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| CCSD | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| CCSD=FULL | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| CCSD(T) | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||||
| CCSD(T)=FULL | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
| 6-311+G(3df,2p) | cc-pVDZ | cc-pVTZ | aug-cc-pVDZ | aug-cc-pVTZ | cc-pV(T+d)Z | ||
|---|---|---|---|---|---|---|---|
| Moller Plesset perturbation | MP2FC// HF/6-31G* | 0.0 b |
|||||
| MP2FC// B3LYP/6-31G* | 0.0 b |
||||||
| MP2FC// MP2FC/6-31G* | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
|||
| MP4// HF/6-31G* | 0.0 b |
||||||
| MP4// B3LYP/6-31G* | 0.0 b |
||||||
| MP4// MP2/6-31G* | 0.0 b |
0.0 b |
|||||
| Coupled Cluster | CCSD// HF/6-31G* | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
| CCSD(T)// HF/6-31G* | 0.0 b |
||||||
| CCSD// B3LYP/6-31G* | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||
| CCSD(T)// B3LYP/6-31G* | 0.0 b |
||||||
| CCSD(T)//B3LYP/6-31G(2df,p) | 0.0 b |
0.0 b |
|||||
| CCSD// MP2FC/6-31G* | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
0.0 b |
||
| CCSD(T)// MP2FC/6-31G* | 0.0 b |
0.0 b |
0.0 b |
0.0 b |
For descriptions of the methods (AM1, HF, MP2, ...) and basis sets (3-21G, 3-21G*, 6-31G, ...) see the glossary in section I.C. Predefined means the basis set used is determined by the method.
gaw refers to the group additivity method implemeted in the NIST Chemistry Webbook.
See section III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.