|
|
IV.A.4. (XIV.F.) |
Relative enthalpies of isomers - Comparison of 298.15K enthalpies (kJ mol-1)
Isomers of C4H7NO
| index | Species | CAS number | Name | Relative experimental enthalpy (kJ mol-1) | sketch |
|---|---|---|---|---|---|
| a | C4H7NO | 616455 | 2-Pyrrolidinone | 0.0 | 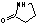 |
| b | C4H7NO | 79390 | Methacrylamide | 39.7 | 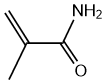 |
| c | C4H7NO | 1120645 | Oxazole, 4,5-dihydro-2-methyl- | 66.9 | 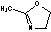 |
| d | C4H7NO | 62957602 | Ethoxyacetonitrile | 127.9 | 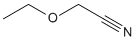 |
| semi-empirical | AM1 | 0.0 a 52.6 b 90.2 c |
|---|---|---|
| PM3 | 0.0 a 69.2 b 75.4 c 430.7 d |
|
| MNDOd | 0.0 a 83.7 b 50.6 c |
|
| composite | G1 | 0.0 a 27.7 b 65.1 c 119.5 d |
| G2MP2 | 0.0 a 29.0 b 68.2 c 122.9 d |
|
| G2 | 0.0 a 28.8 b 68.6 c 79.3 d |
|
| G3 | 0.0 a -61.5 b -19.4 c -10.4 d |
|
| G3B3 | 0.0 a 27.1 b 68.6 c |
|
| G3MP2 | 0.0 a 25.2 b 68.9 c 120.9 d |
|
| G4 | 0.0 a -15.9 b 67.8 c 119.9 d |
|
| CBS-Q | 0.0 a 392.4 d |
|
| molecular mechanics | MM3 | 0.0 a -131.0 b 91.5 c |
| STO-3G | 3-21G | 3-21G* | 6-31G | 6-31G* | 6-31G** | 6-31+G** | 6-311G* | 6-311G** | 6-31G(2df,p) | 6-311+G(3df,2p) | TZVP | cc-pVDZ | cc-pVTZ | aug-cc-pVDZ | aug-cc-pVTZ | cc-pV(T+d)Z | daug-cc-pVTZ | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hartree fock | HF | 0.0 a 184.5 b 0.9 c 88.1 d |
0.0 a 50.7 b 97.6 c 107.9 d |
0.0 a 50.7 b 97.6 c 107.9 d |
0.0 a 27.4 b 109.6 c 128.0 d |
0.0 a 52.7 b 72.1 c 116.1 d |
0.0 a 44.5 b 77.6 c 121.3 d |
0.0 a 39.6 b 79.9 c 121.9 d |
0.0 a 46.5 b 76.8 c 111.6 d |
0.0 a 38.1 b 79.7 c 114.8 d |
0.0 a 43.7 b 73.7 c 117.9 d |
0.0 a 36.5 b 75.5 c |
0.0 a 37.5 b 79.9 c 111.6 d |
0.0 a 39.9 b 77.9 c 118.6 d |
0.0 a 74.9 c 112.7 d |
0.0 a 40.5 b 78.2 c 122.1 d |
0.0 a 35.4 b 74.7 c 112.8 d |
0.0 a 74.9 c |
0.0 a 35.3 b 74.7 c 112.6 d |
| density functional | LSDA | 0.0 a 13.1 c 202.5 d |
0.0 a 106.1 c 214.9 d |
0.0 a 106.1 c 214.9 d |
0.0 a 110.9 c 224.5 d |
0.0 a 75.2 c 207.1 d |
0.0 a 80.5 c 212.1 d |
0.0 a 85.9 c 215.2 d |
0.0 a 88.4 c 206.3 d |
0.0 a 91.5 c 210.0 d |
0.0 a 79.4 c 213.2 d |
NC |
0.0 a 84.7 c 215.2 d |
0.0 a 85.8 c 206.7 d |
0.0 a 87.9 c 218.4 d |
NC |
0.0 a 85.8 c |
||
| BLYP | 0.0 a 135.6 b -12.8 c 108.0 d |
0.0 a 43.9 b 76.6 c 117.5 d |
0.0 a 117.5 d |
0.0 a 18.7 b 83.9 c 129.0 d |
0.0 a 35.8 b 57.1 c 118.1 d |
0.0 a 30.0 b 62.5 c 123.3 d |
0.0 a 23.5 b 68.8 c 126.2 d |
0.0 a 116.1 d |
0.0 a 20.1 b 71.8 c 119.9 d |
0.0 a 29.1 b 62.1 c 123.2 d |
NC |
0.0 a 29.0 b 67.5 c 126.3 d |
0.0 a 16.8 b 68.2 c 116.0 d |
0.0 a 24.2 b 72.4 c |
0.0 a 16.8 b 68.2 c |
||||
| B1B95 | 0.0 a 161.8 b 3.5 c 135.0 d |
0.0 a 66.7 b 95.9 c 167.1 d |
0.0 a 66.7 b 95.9 c 167.1 d |
0.0 a 42.5 b 103.2 c 181.4 d |
0.0 a 63.0 b 69.5 c 154.5 d |
0.0 a 54.5 b 74.1 c 192.6 d |
0.0 a 49.8 b 78.6 c 171.4 d |
0.0 a 55.2 b 77.8 c 162.5 d |
0.0 a 46.9 b 81.0 c 166.3 d |
0.0 a 54.6 b 71.8 c 167.1 d |
NC |
0.0 a 53.4 b 76.5 c 169.0 d |
0.0 a 47.5 b 76.8 c 154.3 d |
0.0 a 51.4 b 80.1 c 164.7 d |
NC |
0.0 a 47.5 b 76.8 c |
|||
| B3LYP | 0.0 a 152.7 b -4.6 c 116.8 d |
0.0 a 53.0 b 87.3 c 131.0 d |
0.0 a 53.0 b 87.3 c 131.0 d |
0.0 a 28.6 b 94.9 c 144.5 d |
0.0 a 47.7 b 63.7 c 131.9 d |
0.0 a 41.2 b 69.2 c 137.2 d |
0.0 a 35.3 b 74.6 c 139.8 d |
0.0 a 39.4 b 74.1 c 129.2 d |
0.0 a 31.5 b 77.3 c 133.0 d |
0.0 a 39.8 b 68.1 c 135.9 d |
0.0 a 28.8 b 74.3 c |
0.0 a 29.8 b 79.1 c 131.9 d |
0.0 a 39.6 b 73.1 c 138.8 d |
0.0 a 28.4 b 73.3 c 129.6 d |
0.0 a 35.1 b 77.1 c 143.0 d |
0.0 a 27.9 b 73.9 c 130.4 d |
0.0 a 28.4 b 73.3 c |
||
| B3LYPultrafine | 0.0 a 47.7 b 63.8 c 132.0 d |
NC |
NC |
NC |
0.0 a 27.9 b 74.0 c 130.4 d |
||||||||||||||
| B3PW91 | 0.0 a 163.6 b 3.2 c 136.7 d |
0.0 a 68.2 b 98.6 c 161.3 d |
0.0 a 68.2 b 98.6 c 161.2 d |
0.0 a 43.7 b 104.1 c 170.9 d |
0.0 a 63.2 b 71.2 c 157.9 d |
0.0 a 56.2 b 76.6 c 163.1 d |
0.0 a 51.7 b 80.8 c 165.4 d |
0.0 a 56.3 b 80.3 c 155.9 d |
0.0 a 47.9 b 159.7 d |
0.0 a 55.3 b 74.3 c 161.4 d |
NC |
0.0 a 55.1 b 78.8 c 163.8 d |
0.0 a 45.9 b 78.6 c 156.1 d |
0.0 a 51.9 b 81.6 c |
0.0 a 45.9 b 78.6 c |
||||
| mPW1PW91 | 0.0 a 169.6 b 4.8 c 141.2 d |
0.0 a 72.7 b 100.8 c 161.5 d |
0.0 a 72.6 b 100.9 c 167.1 d |
0.0 a 47.8 b 106.7 c 176.7 d |
0.0 a 67.8 b 72.8 c 158.2 d |
0.0 a 60.6 b 78.3 c 163.5 d |
0.0 a 56.0 b 82.6 c 165.9 d |
0.0 a 61.1 b 81.5 c 156.0 d |
0.0 a 52.3 b 84.7 c 165.0 d |
0.0 a 59.7 b 75.7 c 166.8 d |
NC |
0.0 a 59.2 b 80.2 c 163.7 d |
0.0 a 45.3 b 74.8 c 156.4 d |
0.0 a 56.2 b 82.9 c 172.4 d |
NC |
0.0 a 45.3 b 74.8 c |
|||
| M06-2X | NC |
NC |
0.0 a 63.7 b 91.5 c 143.2 d |
NC |
0.0 a 57.6 b 64.0 c 135.2 d |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
||||
| PBEPBE | 0.0 a 153.7 b -1.2 c 142.5 d |
0.0 a 66.4 b 91.1 c 161.3 d |
0.0 a 66.4 b 91.1 c 161.3 d |
0.0 a 40.0 b 97.3 c 169.2 d |
0.0 a 58.2 b 67.7 c 157.4 d |
0.0 a 51.5 b 73.1 c 162.7 d |
0.0 a 46.2 b 78.7 c 165.4 d |
0.0 a 51.4 b 77.8 c 156.4 d |
0.0 a 43.1 b 81.2 c 160.3 d |
0.0 a 51.2 b 71.1 c 162.0 d |
NC |
0.0 a 50.7 b 76.4 c 164.5 d |
0.0 a 41.0 b 76.9 c 156.9 d |
0.0 a 46.7 b 80.0 c 168.1 d |
NC |
0.0 a 41.0 b 76.9 c |
|||
| PBEPBEultrafine | 0.0 a 58.2 b 67.7 c 157.4 d |
NC |
NC |
NC |
NC |
||||||||||||||
| PBE1PBE | NC |
NC |
NC |
NC |
0.0 a 70.1 b 73.9 c 167.9 d |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
||||
| HSEh1PBE | NC |
0.0 a 71.9 b 101.4 c 168.4 d |
NC |
NC |
0.0 a 73.4 c 164.8 d |
NC |
0.0 a 55.0 b 83.3 c |
NC |
NC |
NC |
NC |
NC |
0.0 a 49.2 b 80.9 c 162.9 d |
NC |
NC |
||||
| TPSSh | 0.0 a 61.6 b 59.0 c 141.8 d |
0.0 a 50.6 b 68.6 c 149.1 d |
0.0 a 54.5 b 61.7 c 144.3 d |
0.0 a 45.4 b 66.2 c 139.7 d |
|||||||||||||||
| wB97X-D | 0.0 a 67.8 b 108.5 c 164.2 d |
0.0 a 64.0 b 76.1 c 158.2 d |
0.0 a 52.4 b 85.3 c 165.1 d |
0.0 a 48.3 b 88.0 c 158.3 d |
0.0 a 47.6 b 89.3 c 156.8 d |
0.0 a 52.4 b 85.3 c 165.1 d |
0.0 a 46.4 b 82.7 c 154.1 d |
0.0 a 46.3 b 83.1 c 154.7 d |
|||||||||||
| B97D3 | 0.0 a 48.3 b 91.2 c 144.2 d |
0.0 a 41.8 b 67.9 c 141.2 d |
0.0 a 30.1 b 78.1 c 148.8 d |
0.0 a 27.4 b 80.7 c 143.8 d |
0.0 a 24.7 b 77.9 c 142.0 d |
0.0 a 25.3 b 82.3 c 142.1 d |
0.0 a 24.3 b 76.6 c 139.1 d |
0.0 a 23.8 b 77.3 c 140.0 d |
|||||||||||
| STO-3G | 3-21G | 3-21G* | 6-31G | 6-31G* | 6-31G** | 6-31+G** | 6-311G* | 6-311G** | 6-31G(2df,p) | 6-311+G(3df,2p) | TZVP | cc-pVDZ | cc-pVTZ | aug-cc-pVDZ | aug-cc-pVTZ | cc-pV(T+d)Z | daug-cc-pVTZ | ||
| Moller Plesset perturbation | MP2 | 0.0 a 135.5 b -3.0 c -1.3 d |
0.0 a 41.7 b 83.0 c 72.9 d |
0.0 a 41.7 b 83.0 c 72.9 d |
0.0 a 16.5 b 91.6 c 86.7 d |
0.0 a 55.2 b 119.3 d |
0.0 a 50.4 b 75.2 c 122.5 d |
d |
0.0 a 52.3 b 125.4 d |
0.0 a 46.3 b 83.3 c 126.3 d |
0.0 a 53.6 b 76.7 c 135.3 d |
0.0 a 41.0 b 121.1 d |
0.0 a 47.5 b 81.0 c 126.3 d |
d |
0.0 a | ||||
| MP2=FULL | NC |
0.0 a 73.5 d |
0.0 a 73.5 d |
0.0 a 87.2 d |
0.0 a 56.0 b 71.4 c 120.5 d |
0.0 a 123.7 d |
0.0 a 128.2 d |
0.0 a 53.1 b 81.7 c 127.4 d |
0.0 a 47.2 b 84.5 c 128.4 d |
NC |
NC |
0.0 a 127.2 d |
d |
||||||
| MP3 | 0.0 a 52.5 b 64.4 c 125.1 d |
||||||||||||||||||
| MP3=FULL | 0.0 a 53.2 b 65.0 c 126.0 d |
0.0 a 42.6 b 132.0 d |
|||||||||||||||||
| MP4 | NC |
NC |
NC |
NC |
NC |
||||||||||||||
| MP4=FULL | NC |
NC |
NC |
NC |
|||||||||||||||
| B2PLYP | 0.0 a 50.3 b 66.4 c 125.8 d |
0.0 a 33.0 b 74.4 c |
|||||||||||||||||
| Configuration interaction | CID | 0.0 a 99.1 d |
NC |
NC |
0.0 a 130.3 d |
NC |
|||||||||||||
| CISD | 0.0 a 100.1 d |
NC |
NC |
0.0 a 130.0 d |
NC |
||||||||||||||
| STO-3G | 3-21G | 3-21G* | 6-31G | 6-31G* | 6-31G** | 6-31+G** | 6-311G* | 6-311G** | 6-31G(2df,p) | 6-311+G(3df,2p) | TZVP | cc-pVDZ | cc-pVTZ | aug-cc-pVDZ | aug-cc-pVTZ | cc-pV(T+d)Z | daug-cc-pVTZ | ||
| Quadratic configuration interaction | QCISD | 0.0 a 78.3 c 83.1 d |
0.0 a 83.1 d |
0.0 a 100.0 d |
0.0 a 117.4 d |
d |
d |
d |
NC |
NC |
NC |
d |
NC |
||||||
| QCISD(T) | NC |
||||||||||||||||||
| Coupled Cluster | CCD | 0.0 a 77.9 d |
0.0 a 77.9 d |
0.0 a 95.9 d |
0.0 a 115.9 d |
0.0 a 118.3 d |
d |
d |
NC |
NC |
NC |
0.0 a 119.9 d |
NC |
||||||
| CCSD | NC |
||||||||||||||||||
| CCSD=FULL | NC |
||||||||||||||||||
| CCSD(T) | NC |
||||||||||||||||||
| CCSD(T)=FULL | NC |
NC |
NC |
||||||||||||||||
| STO-3G | 3-21G | 3-21G* | 6-31G | 6-31G* | 6-31G** | 6-31+G** | 6-311G* | 6-311G** | 6-31G(2df,p) | 6-311+G(3df,2p) | TZVP | cc-pVDZ | cc-pVTZ | aug-cc-pVDZ | aug-cc-pVTZ | cc-pV(T+d)Z | daug-cc-pVTZ |
| CEP-31G | CEP-31G* | CEP-121G | CEP-121G* | LANL2DZ | SDD | cc-pVTZ-PP | aug-cc-pVTZ-PP | Def2TZVPP | ||
|---|---|---|---|---|---|---|---|---|---|---|
| hartree fock | HF | 0.0 a 42.9 b 102.4 c 134.1 d |
0.0 a 68.2 b 67.9 c 126.6 d |
0.0 a 35.2 b 101.6 c 128.5 d |
0.0 a 52.4 b 69.3 c 111.6 d |
0.0 a 31.9 b 108.0 c 131.8 d |
0.0 a 32.3 b 108.2 c 132.4 d |
0.0 a 35.0 b 76.1 c 113.3 d |
||
| density functional | B1B95 | 0.0 a 61.5 b 102.0 c |
0.0 a 81.2 b 70.2 c |
|||||||
| B3LYP | 0.0 a 42.7 b 89.6 c 153.2 d |
0.0 a 60.6 b 62.2 c 144.9 d |
0.0 a 33.0 b 88.3 c 143.9 d |
0.0 a 45.9 b 63.3 c 129.5 d |
0.0 a 32.1 b 94.6 c 154.3 d |
0.0 a 32.1 b 94.6 c 154.5 d |
0.0 a 28.5 b 75.3 c 131.7 d |
|||
| PBEPBE | 0.0 a 41.0 b 78.7 c 158.8 d |
|||||||||
| Moller Plesset perturbation | MP2 | 0.0 a 27.9 b 88.6 c 88.1 d |
0.0 a 67.2 b 66.5 c 128.8 d |
0.0 a 18.4 b 88.1 c 84.7 d |
0.0 a 51.8 b 67.7 c 120.6 d |
0.0 a 17.4 b 95.4 c 84.2 d |
0.0 a 17.3 b 95.2 c 84.2 d |
0.0 a 43.2 b 131.8 d |
||
| MP2=FULL | NC |
NC |
| cc-pVDZ | cc-pVTZ | aug-cc-pVDZ | cc-pV(T+d)Z | ||
|---|---|---|---|---|---|
| Moller Plesset perturbation | MP2FC// HF/6-31G* | 0.0 a -32.0 b -133.9 c |
0.0 a -40.5 b -123.9 c |
0.0 a -40.5 b -123.9 c |
|
| MP2FC// B3LYP/6-31G* | 0.0 a -52.8 b -19.8 c |
||||
| MP2FC// MP2FC/6-31G* | 0.0 a 41.4 b 81.2 c |
For descriptions of the methods (AM1, HF, MP2, ...) and basis sets (3-21G, 3-21G*, 6-31G, ...) see the glossary in section I.C. Predefined means the basis set used is determined by the method.
gaw refers to the group additivity method implemeted in the NIST Chemistry Webbook.
See section III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.