|
|
IV.A.4. (XIV.F.) |
Relative enthalpies of isomers - Comparison of 298.15K enthalpies (kJ mol-1)
Isomers of CH4O2
| index | Species | CAS number | Name | Relative experimental enthalpy (kJ mol-1) | sketch |
|---|---|---|---|---|---|
| a | H2OH2CO | 55 | water formaldehyde dimer | 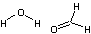 |
|
| b | CH2(OH)2 | 463570 | methanediol |  |
|
| c | CH3OOH | 3031730 | Methyl peroxide | 0.0 | 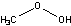 |
| semi-empirical | AM1 | 0.0 c |
|---|---|---|
| PM3 | -222.0 a -122.0 b 0.0 c |
|
| composite | G1 | -248.1 a -256.6 b 0.0 c |
| G2MP2 | -252.8 a -260.7 b 0.0 c |
|
| G2 | -251.8 a -260.6 b 0.0 c |
|
| G3 | -260.3 b 0.0 c |
|
| G3B3 | NC NC |
|
| G3MP2 | -253.6 a 0.0 c |
|
| G4 | -244.1 a -258.5 b 0.0 c |
|
| CBS-Q | NC |
| STO-3G | 3-21G | 3-21G* | 6-31G | 6-31G* | 6-31G** | 6-31+G** | 6-311G* | 6-311G** | 6-31G(2df,p) | 6-311+G(3df,2p) | 6-311+G(3df,2pd) | TZVP | cc-pVDZ | cc-pVTZ | cc-pVQZ | aug-cc-pVDZ | aug-cc-pVTZ | cc-pV(T+d)Z | daug-cc-pVTZ | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hartree fock | HF | 60.6 a -94.5 b 0.0 c |
-160.7 a -216.2 b 0.0 c |
-216.2 b 0.0 c |
-252.7 b 0.0 c |
-239.3 a -267.6 b 0.0 c |
-280.4 b 0.0 c |
-288.5 b 0.0 c |
-269.4 b 0.0 c |
-284.4 b 0.0 c |
NC |
0.0 c |
NC | -262.7 a -281.8 b 0.0 c |
-278.5 b 0.0 c |
-283.3 b 0.0 c |
0.0 c |
-288.9 b 0.0 c |
-266.5 a -286.2 b 0.0 c |
0.0 c |
-266.3 a -286.1 b 0.0 c |
| density functional | LSDA | -86.4 b 0.0 c |
-199.4 b 0.0 c |
-199.4 b 0.0 c |
-224.2 b 0.0 c |
-238.6 b 0.0 c |
-249.0 b 0.0 c |
-260.7 b 0.0 c |
-248.1 b 0.0 c |
-261.3 b 0.0 c |
-249.8 b 0.0 c |
NC |
-251.4 b 0.0 c |
-259.3 b 0.0 c |
-264.3 b 0.0 c |
NC |
0.0 c |
||||
| BLYP | -68.7 b 0.0 c |
-164.4 b 0.0 c |
-164.4 b 0.0 c |
-187.1 b 0.0 c |
-187.4 a -209.8 b 0.0 c |
-220.0 b 0.0 c |
-232.3 b 0.0 c |
-217.3 b 0.0 c |
NC |
NC |
NC |
-221.3 b 0.0 c |
-230.7 b 0.0 c |
0.0 c |
0.0 c |
||||||
| B1B95 | 41.7 a -80.9 b 0.0 c |
-134.1 a -182.9 b 0.0 c |
-134.1 a -182.9 b 0.0 c |
-180.0 a -208.5 b 0.0 c |
-196.6 a -240.3 b 0.0 c |
-207.6 a -206.6 b 0.0 c |
-250.8 b 0.0 c |
-209.0 a -235.9 b 0.0 c |
-224.5 a -249.6 b 0.0 c |
-210.6 a -242.4 b 0.0 c |
NC |
-211.8 a -242.9 b 0.0 c |
-223.1 a -259.2 b 0.0 c |
-228.3 a -265.2 b 0.0 c |
NC |
-223.1 a 0.0 c |
|||||
| B3LYP | -75.9 b 0.0 c |
-134.9 a -181.1 b 0.0 c |
-181.1 b 0.0 c |
-208.2 b 0.0 c |
-195.7 a -229.0 b 0.0 c |
-239.8 b 0.0 c |
-224.8 a -251.5 b 0.0 c |
-236.0 b 0.0 c |
-248.8 b 0.0 c |
-211.2 a -241.4 b 0.0 c |
NC | -229.6 a -250.7 b 0.0 c |
-240.4 b 0.0 c |
-249.6 b 0.0 c |
NC |
-231.4 a -254.2 b 0.0 c |
0.0 c |
||||
| B3LYPultrafine | NC | NC NC |
NC | NC | NC |
-249.5 b 0.0 c |
NC |
-231.4 a -254.2 b 0.0 c |
|||||||||||||
| B3PW91 | -80.9 b 0.0 c |
-190.1 b 0.0 c |
-190.0 b 0.0 c |
-216.8 b 0.0 c |
-235.7 b 0.0 c |
-246.8 b 0.0 c |
-257.1 b 0.0 c |
-243.0 b 0.0 c |
-256.4 b 0.0 c |
NC |
NC |
-247.4 b 0.0 c |
-255.0 b 0.0 c |
0.0 c |
0.0 c |
||||||
| mPW1PW91 | -82.9 b 0.0 c |
-197.5 b 0.0 c |
-197.5 b 0.0 c |
-224.4 b 0.0 c |
-242.6 b 0.0 c |
-253.9 b 0.0 c |
-264.2 b 0.0 c |
-249.5 b 0.0 c |
-260.0 b 0.0 c |
NC |
NC |
-254.6 b 0.0 c |
-261.6 b 0.0 c |
NC |
NC |
0.0 c |
|||||
| M06-2X | NC |
NC |
-136.5 a -203.0 b 0.0 c |
NC |
-197.0 a -246.5 b 0.0 c |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
||||||
| PBEPBE | -77.6 b 0.0 c |
NC NC |
-177.0 b 0.0 c |
NC |
NC NC |
NC |
NC NC |
NC |
-241.0 b 0.0 c |
NC |
NC | NC |
NC |
NC |
NC |
-211.4 a -245.0 b 0.0 c |
|||||
| PBEPBEultrafine | NC | -177.9 a -220.2 b 0.0 c |
NC | NC | NC |
NC |
NC |
NC NC |
|||||||||||||
| PBE1PBE | NC |
NC |
NC |
NC |
-187.3 a -239.4 b 0.0 c |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
NC |
||||||
| HSEh1PBE | NC |
-130.0 a -193.3 b 0.0 c |
NC |
NC |
-188.7 a -238.8 b 0.0 c |
NC |
-216.7 a 0.0 c |
NC |
NC |
NC |
NC |
NC |
-215.5 a -258.0 b 0.0 c |
NC |
NC |
||||||
| TPSSh | -176.0 a 0.0 c |
-203.1 a 0.0 c |
-191.0 a -229.9 b 0.0 c |
-202.9 a 0.0 c |
|||||||||||||||||
| wB97X-D | -139.8 a -196.3 b 0.0 c |
-198.6 a -242.0 b 0.0 c |
-226.0 a -263.6 b 0.0 c |
-227.3 a -261.1 b 0.0 c |
-230.4 a -261.5 b 0.0 c |
-226.0 a -263.6 b 0.0 c |
-226.1 a -259.7 b 0.0 c |
-230.7 a -263.7 b 0.0 c |
|||||||||||||
| B97D3 | -138.1 a -178.9 b 0.0 c |
-197.1 a -223.4 b 0.0 c |
-224.2 a -244.7 b 0.0 c |
-225.1 a -243.4 b 0.0 c |
-233.4 a -250.5 b 0.0 c |
-228.9 a -244.5 b 0.0 c |
-224.4 a -242.9 b 0.0 c |
-230.3 a -247.4 b 0.0 c |
|||||||||||||
| STO-3G | 3-21G | 3-21G* | 6-31G | 6-31G* | 6-31G** | 6-31+G** | 6-311G* | 6-311G** | 6-31G(2df,p) | 6-311+G(3df,2p) | 6-311+G(3df,2pd) | TZVP | cc-pVDZ | cc-pVTZ | cc-pVQZ | aug-cc-pVDZ | aug-cc-pVTZ | cc-pV(T+d)Z | daug-cc-pVTZ | ||
| Moller Plesset perturbation | MP2 | -81.6 b 0.0 c |
-172.6 a -186.7 b 0.0 c |
-186.7 b 0.0 c |
-215.8 b 0.0 c |
-229.1 a -251.0 b 0.0 c |
-260.8 b 0.0 c |
a b |
-237.8 a -256.3 b 0.0 c |
-275.3 b 0.0 c |
NC |
NC | -253.6 a -271.5 b 0.0 c |
-264.2 b 0.0 c |
-273.7 b 0.0 c |
-277.5 b 0.0 c |
NC NC |
0.0 c |
|||
| MP2=FULL | NC |
-186.8 b 0.0 c |
-186.8 b 0.0 c |
-215.8 b 0.0 c |
-251.6 b 0.0 c |
-261.4 b 0.0 c |
-271.8 b 0.0 c |
-257.1 b 0.0 c |
-276.1 b 0.0 c |
NC |
NC |
-264.5 b 0.0 c |
-275.8 b 0.0 c |
NC |
NC |
0.0 c |
|||||
| MP3 | NC |
a b |
|||||||||||||||||||
| MP3=FULL | -218.4 a 0.0 c |
0.0 c |
|||||||||||||||||||
| MP4 | -174.4 b 0.0 c |
-238.0 b 0.0 c |
NC |
NC |
NC |
NC |
NC |
NC |
|||||||||||||
| MP4=FULL | NC |
NC |
NC |
NC |
NC |
NC |
NC |
||||||||||||||
| B2PLYP | -208.5 a 0.0 c |
-233.9 a 0.0 c |
|||||||||||||||||||
| Configuration interaction | CID | NC |
NC |
NC |
-256.4 b 0.0 c |
NC |
|||||||||||||||
| CISD | NC |
NC |
NC |
-254.8 b 0.0 c |
NC |
||||||||||||||||
| STO-3G | 3-21G | 3-21G* | 6-31G | 6-31G* | 6-31G** | 6-31+G** | 6-311G* | 6-311G** | 6-31G(2df,p) | 6-311+G(3df,2p) | 6-311+G(3df,2pd) | TZVP | cc-pVDZ | cc-pVTZ | cc-pVQZ | aug-cc-pVDZ | aug-cc-pVTZ | cc-pV(T+d)Z | daug-cc-pVTZ | ||
| Quadratic configuration interaction | QCISD | -177.5 b 0.0 c |
-177.5 b 0.0 c |
-209.3 b 0.0 c |
-244.4 b 0.0 c |
-253.7 b 0.0 c |
NC |
-247.1 b 0.0 c |
NC |
NC |
NC |
-239.7 a -254.4 b 0.0 c |
NC |
NC |
NC |
||||||
| QCISD(T) | NC |
NC |
NC |
NC |
NC |
NC |
|||||||||||||||
| Coupled Cluster | CCD | -180.3 b 0.0 c |
-180.3 b 0.0 c |
-214.7 b 0.0 c |
-247.6 b 0.0 c |
-256.7 b 0.0 c |
NC |
-249.9 b 0.0 c |
NC |
NC |
NC |
-236.6 a -257.1 b 0.0 c |
NC |
NC |
NC |
||||||
| CCSD | NC |
NC |
NC |
NC |
NC |
NC |
|||||||||||||||
| CCSD=FULL | NC |
NC |
NC |
NC |
NC |
||||||||||||||||
| CCSD(T) | NC | a | NC | NC |
NC |
NC |
NC |
||||||||||||||
| CCSD(T)=FULL | NC |
NC |
NC |
NC |
NC |
||||||||||||||||
| STO-3G | 3-21G | 3-21G* | 6-31G | 6-31G* | 6-31G** | 6-31+G** | 6-311G* | 6-311G** | 6-31G(2df,p) | 6-311+G(3df,2p) | 6-311+G(3df,2pd) | TZVP | cc-pVDZ | cc-pVTZ | cc-pVQZ | aug-cc-pVDZ | aug-cc-pVTZ | cc-pV(T+d)Z | daug-cc-pVTZ |
| CEP-31G | CEP-31G* | CEP-121G | CEP-121G* | LANL2DZ | SDD | cc-pVTZ-PP | aug-cc-pVTZ-PP | Def2TZVPP | ||
|---|---|---|---|---|---|---|---|---|---|---|
| hartree fock | HF | -246.8 b 0.0 c |
-268.8 b 0.0 c |
-248.3 b 0.0 c |
-244.7 a -270.1 b 0.0 c |
-216.4 a -249.9 b 0.0 c |
-249.9 b 0.0 c |
-266.8 a -286.0 b 0.0 c |
||
| density functional | B1B95 | 0.0 c |
-197.0 a 0.0 c |
|||||||
| B3LYP | -209.2 b 0.0 c |
-233.9 b 0.0 c |
-207.6 b 0.0 c |
-201.4 a -233.1 b 0.0 c |
-214.5 b 0.0 c |
-214.4 b 0.0 c |
-230.2 a -253.5 b 0.0 c |
|||
| B3LYPultrafine | NC | |||||||||
| PBEPBE | -209.9 a -244.1 b 0.0 c |
|||||||||
| Moller Plesset perturbation | MP2 | -217.3 b 0.0 c |
-254.6 b 0.0 c |
-218.3 b 0.0 c |
-233.9 a -254.4 b 0.0 c |
-221.7 a -219.8 b 0.0 c |
-219.6 b 0.0 c |
-251.6 a -277.7 b 0.0 c |
| 6-311+G(3df,2p) | cc-pVDZ | cc-pVTZ | aug-cc-pVDZ | cc-pV(T+d)Z | ||
|---|---|---|---|---|---|---|
| Moller Plesset perturbation | MP2FC// HF/6-31G* | -258.2 a 0.0 c |
-250.5 a 0.0 c |
-250.5 a 0.0 c |
||
| MP2FC// B3LYP/6-31G* | -254.1 a 0.0 c |
-248.1 a 0.0 c |
-247.0 a 0.0 c |
-247.0 a 0.0 c |
||
| MP2FC// MP2FC/6-31G* | -247.6 a 0.0 c |
-257.0 a 0.0 c |
-247.6 a 0.0 c |
|||
| MP4// HF/6-31G* | -247.7 a 0.0 c |
-241.8 a 0.0 c |
-241.8 a 0.0 c |
|||
| MP4// B3LYP/6-31G* | -238.9 a 0.0 c |
-237.2 a 0.0 c |
-237.2 a 0.0 c |
|||
| MP4// MP2/6-31G* | -237.9 a 0.0 c |
-237.9 a 0.0 c |
||||
| Coupled Cluster | CCSD// B3LYP/6-31G* | -243.0 a 0.0 c |
||||
| CCSD(T)// B3LYP/6-31G* | -239.4 a 0.0 c |
|||||
| CCSD// MP2FC/6-31G* | -245.1 a 0.0 c |
|||||
| CCSD(T)// MP2FC/6-31G* | -241.7 a 0.0 c |
For descriptions of the methods (AM1, HF, MP2, ...) and basis sets (3-21G, 3-21G*, 6-31G, ...) see the glossary in section I.C. Predefined means the basis set used is determined by the method.
gaw refers to the group additivity method implemeted in the NIST Chemistry Webbook.
See section III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.