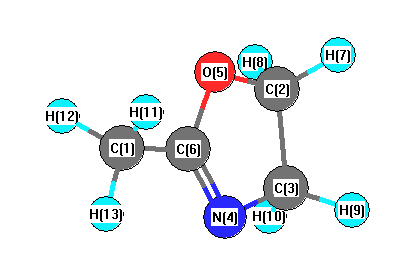
Vibrational Frequencies calculated at HSEh1PBE/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3211 |
3054 |
4.02 |
|
|
|
| 2 |
A |
3097 |
2945 |
48.21 |
|
|
|
| 3 |
A |
3088 |
2937 |
8.18 |
|
|
|
| 4 |
A |
3078 |
2928 |
43.66 |
|
|
|
| 5 |
A |
1790 |
1702 |
146.47 |
|
|
|
| 6 |
A |
1556 |
1480 |
2.03 |
|
|
|
| 7 |
A |
1529 |
1454 |
0.13 |
|
|
|
| 8 |
A |
1504 |
1430 |
9.07 |
|
|
|
| 9 |
A |
1446 |
1375 |
57.80 |
|
|
|
| 10 |
A |
1402 |
1333 |
11.62 |
|
|
|
| 11 |
A |
1358 |
1292 |
13.55 |
|
|
|
| 12 |
A |
1280 |
1217 |
101.86 |
|
|
|
| 13 |
A |
1109 |
1055 |
3.55 |
|
|
|
| 14 |
A |
1061 |
1009 |
92.98 |
|
|
|
| 15 |
A |
1007 |
958 |
0.97 |
|
|
|
| 16 |
A |
969 |
922 |
18.75 |
|
|
|
| 17 |
A |
933 |
887 |
18.23 |
|
|
|
| 18 |
A |
768 |
731 |
5.55 |
|
|
|
| 19 |
A |
675 |
641 |
2.74 |
|
|
|
| 20 |
A |
350 |
333 |
5.85 |
|
|
|
| 21 |
A |
3156 |
3001 |
6.90 |
|
|
|
| 22 |
A |
3148 |
2994 |
43.21 |
|
|
|
| 23 |
A |
3112 |
2959 |
8.08 |
|
|
|
| 24 |
A |
1501 |
1427 |
9.98 |
|
|
|
| 25 |
A |
1264 |
1203 |
0.05 |
|
|
|
| 26 |
A |
1234 |
1174 |
1.39 |
|
|
|
| 27 |
A |
1134 |
1079 |
0.01 |
|
|
|
| 28 |
A |
1075 |
1022 |
4.50 |
|
|
|
| 29 |
A |
841 |
800 |
0.24 |
|
|
|
| 30 |
A |
600 |
570 |
5.71 |
|
|
|
| 31 |
A |
240 |
228 |
10.51 |
|
|
|
| 32 |
A |
142 |
135 |
1.09 |
|
|
|
| 33 |
A |
73 |
70 |
0.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24365.4 cm
-1
Scaled (by 0.951) Zero Point Vibrational Energy (zpe) 23171.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.587 |
|
|
|
| 2 |
C |
-0.108 |
|
|
|
| 3 |
C |
-0.240 |
|
|
|
| 4 |
N |
-0.449 |
|
|
|
| 5 |
O |
-0.481 |
|
|
|
| 6 |
C |
0.519 |
|
|
|
| 7 |
H |
0.181 |
|
|
|
| 8 |
H |
0.181 |
|
|
|
| 9 |
H |
0.184 |
|
|
|
| 10 |
H |
0.184 |
|
|
|
| 11 |
H |
0.202 |
|
|
|
| 12 |
H |
0.202 |
|
|
|
| 13 |
H |
0.212 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.993 |
-0.892 |
0.000 |
1.335 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.506 |
1.776 |
0.000 |
| y |
1.776 |
-29.305 |
0.000 |
| z |
0.000 |
0.000 |
-34.857 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-8.424 |
1.776 |
0.000 |
| y |
1.776 |
8.376 |
0.000 |
| z |
0.000 |
0.000 |
0.048 |
|
| Polar |
|---|
| 3z2-r2 | 0.096 |
|---|
| x2-y2 | -11.200 |
|---|
| xy | 1.776 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.291 |
0.682 |
0.000 |
| y |
0.682 |
8.739 |
0.000 |
| z |
0.000 |
0.000 |
5.510 |
<r2> (average value of r
2) Å
2
| <r2> |
147.528 |
| (<r2>)1/2 |
12.146 |