Jump to
S1C2
Energy calculated at PBE1PBE/6-31G*
| | hartrees |
|---|
| Energy at 0K | -323.274968 |
| Energy at 298.15K | -323.284033 |
| HF Energy | -323.274968 |
| Nuclear repulsion energy | 242.379382 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBE1PBE/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3164 |
3007 |
23.85 |
|
|
|
| 2 |
A |
3154 |
2998 |
34.10 |
|
|
|
| 3 |
A |
3141 |
2985 |
20.77 |
|
|
|
| 4 |
A |
3117 |
2962 |
13.11 |
|
|
|
| 5 |
A |
3081 |
2927 |
27.18 |
|
|
|
| 6 |
A |
3076 |
2923 |
23.11 |
|
|
|
| 7 |
A |
3073 |
2920 |
11.13 |
|
|
|
| 8 |
A |
1813 |
1723 |
231.48 |
|
|
|
| 9 |
A |
1534 |
1457 |
9.29 |
|
|
|
| 10 |
A |
1527 |
1451 |
9.46 |
|
|
|
| 11 |
A |
1511 |
1436 |
1.78 |
|
|
|
| 12 |
A |
1504 |
1429 |
2.92 |
|
|
|
| 13 |
A |
1443 |
1372 |
7.61 |
|
|
|
| 14 |
A |
1421 |
1351 |
11.16 |
|
|
|
| 15 |
A |
1397 |
1327 |
2.14 |
|
|
|
| 16 |
A |
1322 |
1256 |
4.99 |
|
|
|
| 17 |
A |
1307 |
1242 |
10.50 |
|
|
|
| 18 |
A |
1204 |
1144 |
9.93 |
|
|
|
| 19 |
A |
1135 |
1079 |
13.87 |
|
|
|
| 20 |
A |
1115 |
1060 |
27.09 |
|
|
|
| 21 |
A |
1027 |
976 |
7.08 |
|
|
|
| 22 |
A |
953 |
905 |
188.85 |
|
|
|
| 23 |
A |
896 |
852 |
7.69 |
|
|
|
| 24 |
A |
887 |
843 |
266.94 |
|
|
|
| 25 |
A |
794 |
755 |
7.05 |
|
|
|
| 26 |
A |
640 |
608 |
15.35 |
|
|
|
| 27 |
A |
463 |
440 |
4.07 |
|
|
|
| 28 |
A |
390 |
370 |
3.33 |
|
|
|
| 29 |
A |
287 |
273 |
0.58 |
|
|
|
| 30 |
A |
245 |
233 |
0.78 |
|
|
|
| 31 |
A |
186 |
177 |
0.45 |
|
|
|
| 32 |
A |
108 |
103 |
0.83 |
|
|
|
| 33 |
A |
58 |
55 |
0.02 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23487.2 cm
-1
Scaled (by 0.9503) Zero Point Vibrational Energy (zpe) 22319.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBE1PBE/6-31G*
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| H1 |
0.553 |
-2.109 |
0.121 |
| N2 |
-1.500 |
0.139 |
0.423 |
| O3 |
-2.434 |
0.545 |
-0.168 |
| O4 |
-0.636 |
-0.552 |
-0.426 |
| C5 |
1.769 |
-0.340 |
-0.202 |
| H6 |
1.883 |
-0.536 |
-1.275 |
| H7 |
2.622 |
-0.820 |
0.295 |
| C8 |
0.508 |
-1.028 |
0.290 |
| H9 |
0.341 |
-0.844 |
1.358 |
| H10 |
2.705 |
1.616 |
-0.297 |
| H11 |
1.691 |
1.375 |
1.134 |
| H12 |
0.944 |
1.654 |
-0.447 |
| C13 |
1.778 |
1.160 |
0.063 |
Atom - Atom Distances (Å)
| |
H1 |
N2 |
O3 |
O4 |
C5 |
H6 |
H7 |
C8 |
H9 |
H10 |
H11 |
H12 |
C13 |
| H1 | | 3.0594 | 4.0066 | 2.0337 | 2.1712 | 2.4888 | 2.4441 | 1.0950 | 1.7821 | 4.3228 | 3.8028 | 3.8255 | 3.4912 |
N2 | 3.0594 | | 1.1774 | 1.3944 | 3.3628 | 3.8450 | 4.2344 | 2.3266 | 2.2875 | 4.5153 | 3.4956 | 3.0043 | 3.4521 | O3 | 4.0066 | 1.1774 | | 2.1225 | 4.2956 | 4.5860 | 5.2578 | 3.3678 | 3.4588 | 5.2514 | 4.4046 | 3.5663 | 4.2626 | O4 | 2.0337 | 1.3944 | 2.1225 | | 2.4247 | 2.6583 | 3.3476 | 1.4306 | 2.0547 | 3.9854 | 3.4004 | 2.7135 | 2.9992 | C5 | 2.1712 | 3.3628 | 4.2956 | 2.4247 | | 1.0970 | 1.0981 | 1.5184 | 2.1740 | 2.1707 | 2.1755 | 2.1714 | 1.5227 | H6 | 2.4888 | 3.8450 | 4.5860 | 2.6583 | 1.0970 | | 1.7592 | 2.1404 | 3.0669 | 2.5033 | 3.0813 | 2.5223 | 2.1625 | H7 | 2.4441 | 4.2344 | 5.2578 | 3.3476 | 1.0981 | 1.7592 | | 2.1242 | 2.5161 | 2.5087 | 2.5277 | 3.0800 | 2.1648 | C8 | 1.0950 | 2.3266 | 3.3678 | 1.4306 | 1.5184 | 2.1404 | 2.1242 | | 1.0971 | 3.4877 | 2.8085 | 2.8149 | 2.5396 | H9 | 1.7821 | 2.2875 | 3.4588 | 2.0547 | 2.1740 | 3.0669 | 2.5161 | 1.0971 | | 3.7918 | 2.6067 | 3.1397 | 2.7847 | H10 | 4.3228 | 4.5153 | 5.2514 | 3.9854 | 2.1707 | 2.5033 | 2.5087 | 3.4877 | 3.7918 | | 1.7703 | 1.7680 | 1.0946 | H11 | 3.8028 | 3.4956 | 4.4046 | 3.4004 | 2.1755 | 3.0813 | 2.5277 | 2.8085 | 2.6067 | 1.7703 | | 1.7705 | 1.0963 | H12 | 3.8255 | 3.0043 | 3.5663 | 2.7135 | 2.1714 | 2.5223 | 3.0800 | 2.8149 | 3.1397 | 1.7680 | 1.7705 | | 1.0948 | C13 | 3.4912 | 3.4521 | 4.2626 | 2.9992 | 1.5227 | 2.1625 | 2.1648 | 2.5396 | 2.7847 | 1.0946 | 1.0963 | 1.0948 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
137.438 |
|
C1 |
C2 |
H10 |
66.399 |
| C1 |
C2 |
H11 |
70.563 |
|
C2 |
C1 |
H7 |
99.999 |
| C2 |
C1 |
H8 |
39.823 |
|
C2 |
C1 |
H9 |
47.953 |
| C2 |
C3 |
O4 |
37.847 |
|
C2 |
C3 |
H12 |
52.789 |
| C2 |
C3 |
H13 |
40.444 |
|
C3 |
C2 |
H10 |
123.077 |
| C3 |
C2 |
H11 |
134.752 |
|
C3 |
O4 |
N5 |
141.613 |
| O4 |
C3 |
H12 |
49.360 |
|
O4 |
C3 |
H13 |
40.885 |
| O4 |
N5 |
O6 |
89.826 |
|
H7 |
C1 |
H8 |
60.197 |
| H7 |
C1 |
H9 |
71.121 |
|
H8 |
C1 |
H9 |
35.650 |
| H10 |
C2 |
H11 |
20.988 |
|
H12 |
C3 |
H13 |
12.438 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBE1PBE/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.188 |
|
|
|
| 2 |
N |
0.292 |
|
|
|
| 3 |
O |
-0.253 |
|
|
|
| 4 |
O |
-0.350 |
|
|
|
| 5 |
C |
-0.338 |
|
|
|
| 6 |
H |
0.188 |
|
|
|
| 7 |
H |
0.175 |
|
|
|
| 8 |
C |
-0.101 |
|
|
|
| 9 |
H |
0.184 |
|
|
|
| 10 |
H |
0.182 |
|
|
|
| 11 |
H |
0.175 |
|
|
|
| 12 |
H |
0.188 |
|
|
|
| 13 |
C |
-0.530 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.466 |
-0.907 |
0.714 |
2.723 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.348 |
0.497 |
-0.619 |
| y |
0.497 |
-34.433 |
-0.841 |
| z |
-0.619 |
-0.841 |
-36.482 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.890 |
0.497 |
-0.619 |
| y |
0.497 |
2.982 |
-0.841 |
| z |
-0.619 |
-0.841 |
-0.092 |
|
| Polar |
|---|
| 3z2-r2 | -0.184 |
|---|
| x2-y2 | -3.915 |
|---|
| xy | 0.497 |
|---|
| xz | -0.619 |
|---|
| yz | -0.841 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.796 |
-1.095 |
0.017 |
| y |
-1.095 |
6.521 |
-0.018 |
| z |
0.017 |
-0.018 |
5.676 |
<r2> (average value of r
2) Å
2
| <r2> |
192.601 |
| (<r2>)1/2 |
13.878 |
Jump to
S1C1
Energy calculated at PBE1PBE/6-31G*
| | hartrees |
|---|
| Energy at 0K | -323.274967 |
| Energy at 298.15K | -323.284032 |
| HF Energy | -323.274967 |
| Nuclear repulsion energy | 242.384085 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBE1PBE/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3164 |
3007 |
23.84 |
|
|
|
| 2 |
A |
3154 |
2998 |
34.12 |
|
|
|
| 3 |
A |
3141 |
2985 |
20.68 |
|
|
|
| 4 |
A |
3117 |
2962 |
13.15 |
|
|
|
| 5 |
A |
3081 |
2928 |
27.11 |
|
|
|
| 6 |
A |
3076 |
2923 |
23.05 |
|
|
|
| 7 |
A |
3073 |
2920 |
11.26 |
|
|
|
| 8 |
A |
1813 |
1723 |
231.58 |
|
|
|
| 9 |
A |
1534 |
1457 |
9.31 |
|
|
|
| 10 |
A |
1527 |
1451 |
9.47 |
|
|
|
| 11 |
A |
1511 |
1436 |
1.77 |
|
|
|
| 12 |
A |
1504 |
1429 |
2.92 |
|
|
|
| 13 |
A |
1443 |
1371 |
7.60 |
|
|
|
| 14 |
A |
1421 |
1351 |
11.14 |
|
|
|
| 15 |
A |
1397 |
1327 |
2.15 |
|
|
|
| 16 |
A |
1322 |
1256 |
4.98 |
|
|
|
| 17 |
A |
1307 |
1242 |
10.53 |
|
|
|
| 18 |
A |
1204 |
1144 |
9.94 |
|
|
|
| 19 |
A |
1135 |
1079 |
13.63 |
|
|
|
| 20 |
A |
1115 |
1060 |
27.22 |
|
|
|
| 21 |
A |
1027 |
976 |
7.11 |
|
|
|
| 22 |
A |
952 |
905 |
188.14 |
|
|
|
| 23 |
A |
896 |
852 |
7.34 |
|
|
|
| 24 |
A |
887 |
843 |
267.82 |
|
|
|
| 25 |
A |
794 |
754 |
7.13 |
|
|
|
| 26 |
A |
640 |
608 |
15.37 |
|
|
|
| 27 |
A |
463 |
440 |
4.09 |
|
|
|
| 28 |
A |
390 |
370 |
3.32 |
|
|
|
| 29 |
A |
287 |
273 |
0.58 |
|
|
|
| 30 |
A |
245 |
233 |
0.78 |
|
|
|
| 31 |
A |
186 |
177 |
0.44 |
|
|
|
| 32 |
A |
108 |
103 |
0.83 |
|
|
|
| 33 |
A |
58 |
55 |
0.02 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23486.8 cm
-1
Scaled (by 0.9503) Zero Point Vibrational Energy (zpe) 22319.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBE1PBE/6-31G*
Point Group is C1
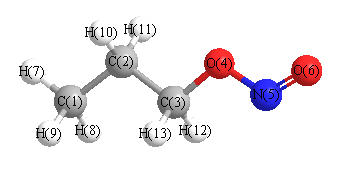
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.181 |
-0.919 |
0.183 |
| C2 |
-1.752 |
0.458 |
-0.313 |
| C3 |
-0.403 |
0.894 |
0.221 |
| O4 |
0.574 |
-0.028 |
-0.289 |
| N5 |
1.788 |
0.207 |
0.316 |
| O6 |
2.562 |
-0.558 |
-0.145 |
| H7 |
-3.164 |
-1.189 |
-0.216 |
| H8 |
-2.245 |
-0.941 |
1.277 |
| H9 |
-1.466 |
-1.686 |
-0.129 |
| H10 |
-1.718 |
0.474 |
-1.409 |
| H11 |
-2.485 |
1.216 |
-0.008 |
| H12 |
-0.141 |
1.903 |
-0.116 |
| H13 |
-0.375 |
0.867 |
1.316 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5250 | 2.5399 | 2.9339 | 4.1278 | 4.7684 | 1.0946 | 1.0963 | 1.0939 | 2.1652 | 2.1654 | 3.4950 | 2.7815 |
C2 | 1.5250 | | 1.5155 | 2.3768 | 3.6045 | 4.4358 | 2.1711 | 2.1741 | 2.1704 | 1.0966 | 1.0975 | 2.1733 | 2.1723 | C3 | 2.5399 | 1.5155 | | 1.4366 | 2.2979 | 3.3216 | 3.4862 | 2.8062 | 2.8120 | 2.1363 | 2.1191 | 1.0954 | 1.0960 | O4 | 2.9339 | 2.3768 | 1.4366 | | 1.3763 | 2.0627 | 3.9151 | 3.3515 | 2.6335 | 2.6004 | 3.3140 | 2.0664 | 2.0683 | N5 | 4.1278 | 3.6045 | 2.2979 | 1.3763 | | 1.1818 | 5.1725 | 4.3018 | 3.7904 | 3.9167 | 4.4021 | 2.6047 | 2.4729 | O6 | 4.7684 | 4.4358 | 3.3216 | 2.0627 | 1.1818 | | 5.7617 | 5.0277 | 4.1831 | 4.5812 | 5.3515 | 3.6560 | 3.5767 | H7 | 1.0946 | 2.1711 | 3.4862 | 3.9151 | 5.1725 | 5.7617 | | 1.7706 | 1.7719 | 2.5055 | 2.5077 | 4.3251 | 3.7884 | H8 | 1.0963 | 2.1741 | 2.8062 | 3.3515 | 4.3018 | 5.0277 | 1.7706 | | 1.7719 | 3.0810 | 2.5223 | 3.8015 | 2.6011 | H9 | 1.0939 | 2.1704 | 2.8120 | 2.6335 | 3.7904 | 4.1831 | 1.7719 | 1.7719 | | 2.5229 | 3.0781 | 3.8254 | 3.1298 | H10 | 2.1652 | 1.0966 | 2.1363 | 2.6004 | 3.9167 | 4.5812 | 2.5055 | 3.0810 | 2.5229 | | 1.7608 | 2.4906 | 3.0636 | H11 | 2.1654 | 1.0975 | 2.1191 | 3.3140 | 4.4021 | 5.3515 | 2.5077 | 2.5223 | 3.0781 | 1.7608 | | 2.4443 | 2.5153 | H12 | 3.4950 | 2.1733 | 1.0954 | 2.0664 | 2.6047 | 3.6560 | 4.3251 | 3.8015 | 3.8254 | 2.4906 | 2.4443 | | 1.7826 | H13 | 2.7815 | 2.1723 | 1.0960 | 2.0683 | 2.4729 | 3.5767 | 3.7884 | 2.6011 | 3.1298 | 3.0636 | 2.5153 | 1.7826 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
113.306 |
|
C1 |
C2 |
H10 |
110.291 |
| C1 |
C2 |
H11 |
110.256 |
|
C2 |
C1 |
H7 |
110.872 |
| C2 |
C1 |
H8 |
111.015 |
|
C2 |
C1 |
H9 |
110.867 |
| C2 |
C3 |
O4 |
107.212 |
|
C2 |
C3 |
H12 |
111.688 |
| C2 |
C3 |
H13 |
111.564 |
|
C3 |
C2 |
H10 |
108.682 |
| C3 |
C2 |
H11 |
107.305 |
|
C3 |
O4 |
N5 |
109.536 |
| O4 |
C3 |
H12 |
108.650 |
|
O4 |
C3 |
H13 |
108.761 |
| O4 |
N5 |
O6 |
107.238 |
|
H7 |
C1 |
H8 |
107.835 |
| H7 |
C1 |
H9 |
108.123 |
|
H8 |
C1 |
H9 |
108.002 |
| H10 |
C2 |
H11 |
106.746 |
|
H12 |
C3 |
H13 |
108.875 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBE1PBE/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.530 |
|
|
|
| 2 |
C |
-0.338 |
|
|
|
| 3 |
C |
-0.101 |
|
|
|
| 4 |
O |
-0.350 |
|
|
|
| 5 |
N |
0.292 |
|
|
|
| 6 |
O |
-0.253 |
|
|
|
| 7 |
H |
0.182 |
|
|
|
| 8 |
H |
0.175 |
|
|
|
| 9 |
H |
0.188 |
|
|
|
| 10 |
H |
0.188 |
|
|
|
| 11 |
H |
0.175 |
|
|
|
| 12 |
H |
0.188 |
|
|
|
| 13 |
H |
0.184 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.466 |
-0.906 |
0.714 |
2.723 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.344 |
0.495 |
-0.620 |
| y |
0.495 |
-34.434 |
-0.842 |
| z |
-0.620 |
-0.842 |
-36.482 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.886 |
0.495 |
-0.620 |
| y |
0.495 |
2.979 |
-0.842 |
| z |
-0.620 |
-0.842 |
-0.093 |
|
| Polar |
|---|
| 3z2-r2 | -0.186 |
|---|
| x2-y2 | -3.910 |
|---|
| xy | 0.495 |
|---|
| xz | -0.620 |
|---|
| yz | -0.842 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.795 |
-1.096 |
0.017 |
| y |
-1.096 |
6.522 |
-0.017 |
| z |
0.017 |
-0.017 |
5.676 |
<r2> (average value of r
2) Å
2
| <r2> |
192.562 |
| (<r2>)1/2 |
13.877 |