Jump to
S1C2
Energy calculated at HF/6-311+G(3df,2p)
| | hartrees |
|---|
| Energy at 0K | -150.836745 |
| Energy at 298.15K | -150.839143 |
| HF Energy | -150.836745 |
| Nuclear repulsion energy | 38.311253 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-311+G(3df,2p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
4154 |
3775 |
30.71 |
79.93 |
0.12 |
0.22 |
| 2 |
A |
1613 |
1466 |
0.31 |
3.33 |
0.45 |
0.62 |
| 3 |
A |
1167 |
1060 |
0.77 |
26.55 |
0.26 |
0.41 |
| 4 |
A |
435 |
395 |
190.51 |
1.06 |
0.73 |
0.85 |
| 5 |
B |
4154 |
3775 |
95.25 |
25.96 |
0.75 |
0.86 |
| 6 |
B |
1503 |
1365 |
103.90 |
0.87 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 6513.1 cm
-1
Scaled (by 0.9086) Zero Point Vibrational Energy (zpe) 5917.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-311+G(3df,2p)
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.691 |
-0.059 |
| O2 |
0.000 |
-0.691 |
-0.059 |
| H3 |
0.750 |
0.906 |
0.469 |
| H4 |
-0.750 |
-0.906 |
0.469 |
Atom - Atom Distances (Å)
| |
O1 |
O2 |
H3 |
H4 |
| O1 | | 1.3825 | 0.9416 | 1.8418 |
O2 | 1.3825 | | 1.8418 | 0.9416 | H3 | 0.9416 | 1.8418 | | 2.3527 | H4 | 1.8418 | 0.9416 | 2.3527 | |
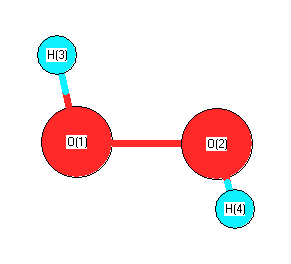 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
O2 |
H4 |
103.196 |
|
O2 |
O1 |
H3 |
103.196 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-311+G(3df,2p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.228 |
|
|
|
| 2 |
O |
-0.228 |
|
|
|
| 3 |
H |
0.228 |
|
|
|
| 4 |
H |
0.228 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.917 |
1.917 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-9.704 |
3.020 |
0.000 |
| y |
3.020 |
-11.208 |
0.000 |
| z |
0.000 |
0.000 |
-11.521 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.660 |
3.020 |
0.000 |
| y |
3.020 |
-0.595 |
0.000 |
| z |
0.000 |
0.000 |
-1.065 |
|
| Polar |
|---|
| 3z2-r2 | -2.130 |
|---|
| x2-y2 | 1.504 |
|---|
| xy | 3.020 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.638 |
0.233 |
0.000 |
| y |
0.233 |
2.441 |
0.000 |
| z |
0.000 |
0.000 |
1.513 |
<r2> (average value of r
2) Å
2
| <r2> |
17.659 |
| (<r2>)1/2 |
4.202 |
Jump to
S1C1
Energy calculated at HF/6-311+G(3df,2p)
| | hartrees |
|---|
| Energy at 0K | -150.834614 |
| Energy at 298.15K | |
| HF Energy | -150.834614 |
| Nuclear repulsion energy | 38.179345 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/6-311+G(3df,2p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
4180 |
3798 |
0.00 |
|
|
|
| 2 |
Ag |
1718 |
1561 |
0.00 |
|
|
|
| 3 |
Ag |
1164 |
1057 |
0.00 |
|
|
|
| 4 |
Au |
325i |
295i |
301.87 |
|
|
|
| 5 |
Bu |
4189 |
3807 |
174.24 |
|
|
|
| 6 |
Bu |
1386 |
1259 |
135.93 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 6156.0 cm
-1
Scaled (by 0.9086) Zero Point Vibrational Energy (zpe) 5593.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/6-311+G(3df,2p)
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.696 |
0.000 |
| O2 |
0.000 |
-0.696 |
0.000 |
| H3 |
0.921 |
0.885 |
0.000 |
| H4 |
-0.921 |
-0.885 |
0.000 |
Atom - Atom Distances (Å)
| |
O1 |
O2 |
H3 |
H4 |
| O1 | | 1.3913 | 0.9406 | 1.8295 |
O2 | 1.3913 | | 1.8295 | 0.9406 | H3 | 0.9406 | 1.8295 | | 2.5550 | H4 | 1.8295 | 0.9406 | 2.5550 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
O2 |
H4 |
101.610 |
|
O2 |
O1 |
H3 |
101.610 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-311+G(3df,2p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.238 |
|
|
|
| 2 |
O |
-0.238 |
|
|
|
| 3 |
H |
0.238 |
|
|
|
| 4 |
H |
0.238 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-8.087 |
3.649 |
0.000 |
| y |
3.649 |
-11.358 |
0.000 |
| z |
0.000 |
0.000 |
-12.825 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.005 |
3.649 |
0.000 |
| y |
3.649 |
-0.903 |
0.000 |
| z |
0.000 |
0.000 |
-3.102 |
|
| Polar |
|---|
| 3z2-r2 | -6.205 |
|---|
| x2-y2 | 3.272 |
|---|
| xy | 3.649 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.741 |
0.263 |
0.000 |
| y |
0.263 |
2.463 |
0.000 |
| z |
0.000 |
0.000 |
1.414 |
<r2> (average value of r
2) Å
2
| <r2> |
17.725 |
| (<r2>)1/2 |
4.210 |