Vibrational Frequencies calculated at HF/CEP-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3960 |
3556 |
41.67 |
|
|
|
| 2 |
A |
3826 |
3436 |
44.20 |
|
|
|
| 3 |
A |
3326 |
2987 |
58.16 |
|
|
|
| 4 |
A |
3299 |
2963 |
62.70 |
|
|
|
| 5 |
A |
3272 |
2938 |
29.24 |
|
|
|
| 6 |
A |
3235 |
2905 |
31.35 |
|
|
|
| 7 |
A |
3224 |
2895 |
56.08 |
|
|
|
| 8 |
A |
1957 |
1757 |
437.96 |
|
|
|
| 9 |
A |
1785 |
1603 |
115.62 |
|
|
|
| 10 |
A |
1629 |
1462 |
11.79 |
|
|
|
| 11 |
A |
1619 |
1454 |
4.38 |
|
|
|
| 12 |
A |
1607 |
1443 |
2.68 |
|
|
|
| 13 |
A |
1567 |
1408 |
29.10 |
|
|
|
| 14 |
A |
1530 |
1374 |
41.01 |
|
|
|
| 15 |
A |
1397 |
1255 |
0.73 |
|
|
|
| 16 |
A |
1379 |
1238 |
159.11 |
|
|
|
| 17 |
A |
1222 |
1098 |
5.30 |
|
|
|
| 18 |
A |
1198 |
1075 |
1.03 |
|
|
|
| 19 |
A |
1148 |
1031 |
9.70 |
|
|
|
| 20 |
A |
1073 |
963 |
2.30 |
|
|
|
| 21 |
A |
875 |
786 |
12.48 |
|
|
|
| 22 |
A |
857 |
769 |
1.99 |
|
|
|
| 23 |
A |
667 |
599 |
12.21 |
|
|
|
| 24 |
A |
637 |
572 |
38.78 |
|
|
|
| 25 |
A |
523 |
470 |
15.59 |
|
|
|
| 26 |
A |
466 |
419 |
6.43 |
|
|
|
| 27 |
A |
357 |
321 |
294.49 |
|
|
|
| 28 |
A |
268 |
241 |
10.35 |
|
|
|
| 29 |
A |
232 |
208 |
0.28 |
|
|
|
| 30 |
A |
33 |
30 |
2.22 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24082.8 cm
-1
Scaled (by 0.898) Zero Point Vibrational Energy (zpe) 21626.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
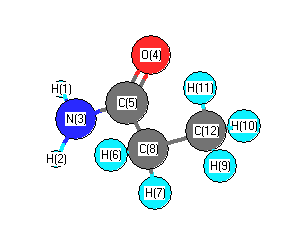
Charges from optimized geometry at HF/CEP-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.371 |
|
|
|
| 2 |
H |
0.357 |
|
|
|
| 3 |
N |
-0.539 |
|
|
|
| 4 |
O |
-0.254 |
|
|
|
| 5 |
C |
-0.058 |
|
|
|
| 6 |
H |
0.100 |
|
|
|
| 7 |
H |
0.104 |
|
|
|
| 8 |
C |
-0.102 |
|
|
|
| 9 |
H |
0.124 |
|
|
|
| 10 |
H |
0.113 |
|
|
|
| 11 |
H |
0.134 |
|
|
|
| 12 |
C |
-0.350 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.684 |
-3.896 |
0.616 |
4.003 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.618 |
2.434 |
-1.240 |
| y |
2.434 |
-33.720 |
-0.804 |
| z |
-1.240 |
-0.804 |
-31.385 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.934 |
2.434 |
-1.240 |
| y |
2.434 |
-5.218 |
-0.804 |
| z |
-1.240 |
-0.804 |
-1.716 |
|
| Polar |
|---|
| 3z2-r2 | -3.431 |
|---|
| x2-y2 | 8.101 |
|---|
| xy | 2.434 |
|---|
| xz | -1.240 |
|---|
| yz | -0.804 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.438 |
0.309 |
0.056 |
| y |
0.309 |
6.207 |
0.017 |
| z |
0.056 |
0.017 |
4.414 |
<r2> (average value of r
2) Å
2
| <r2> |
107.536 |
| (<r2>)1/2 |
10.370 |