Jump to
S1C2
Energy calculated at HF/CEP-121G
| | hartrees |
|---|
| Energy at 0K | -69.347325 |
| Energy at 298.15K | -69.352779 |
| Nuclear repulsion energy | 124.830172 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/CEP-121G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3995 |
3645 |
114.05 |
|
|
|
| 2 |
A' |
3966 |
3619 |
76.78 |
|
|
|
| 3 |
A' |
3807 |
3474 |
82.64 |
|
|
|
| 4 |
A' |
1888 |
1723 |
11.49 |
|
|
|
| 5 |
A' |
1831 |
1671 |
866.79 |
|
|
|
| 6 |
A' |
1796 |
1639 |
112.16 |
|
|
|
| 7 |
A' |
1547 |
1412 |
1.58 |
|
|
|
| 8 |
A' |
1411 |
1288 |
151.33 |
|
|
|
| 9 |
A' |
1239 |
1131 |
191.75 |
|
|
|
| 10 |
A' |
1204 |
1099 |
159.20 |
|
|
|
| 11 |
A' |
830 |
757 |
4.54 |
|
|
|
| 12 |
A' |
638 |
582 |
108.14 |
|
|
|
| 13 |
A' |
572 |
522 |
0.89 |
|
|
|
| 14 |
A' |
438 |
400 |
7.05 |
|
|
|
| 15 |
A' |
287 |
262 |
15.81 |
|
|
|
| 16 |
A" |
886 |
809 |
0.57 |
|
|
|
| 17 |
A" |
704 |
643 |
660.74 |
|
|
|
| 18 |
A" |
647 |
591 |
3.45 |
|
|
|
| 19 |
A" |
620 |
566 |
38.47 |
|
|
|
| 20 |
A" |
456 |
416 |
16.80 |
|
|
|
| 21 |
A" |
52 |
47 |
10.19 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14407.4 cm
-1
Scaled (by 0.9125) Zero Point Vibrational Energy (zpe) 13146.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/CEP-121G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.753 |
0.000 |
| C2 |
-0.052 |
-0.784 |
0.000 |
| O3 |
-1.112 |
-1.403 |
0.000 |
| O4 |
1.058 |
1.365 |
0.000 |
| O5 |
-1.206 |
1.340 |
0.000 |
| N6 |
1.186 |
-1.344 |
0.000 |
| H7 |
1.261 |
-2.335 |
0.000 |
| H8 |
2.004 |
-0.780 |
0.000 |
| H9 |
-1.175 |
2.296 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5380 | 2.4251 | 1.2223 | 1.3411 | 2.4090 | 3.3350 | 2.5228 | 1.9399 |
C2 | 1.5380 | | 1.2267 | 2.4192 | 2.4172 | 1.3586 | 2.0316 | 2.0556 | 3.2788 | O3 | 2.4251 | 1.2267 | | 3.5167 | 2.7439 | 2.2981 | 2.5490 | 3.1767 | 3.6992 | O4 | 1.2223 | 2.4192 | 3.5167 | | 2.2638 | 2.7124 | 3.7055 | 2.3448 | 2.4194 | O5 | 1.3411 | 2.4172 | 2.7439 | 2.2638 | | 3.5949 | 4.4256 | 3.8464 | 0.9568 | N6 | 2.4090 | 1.3586 | 2.2981 | 2.7124 | 3.5949 | | 0.9933 | 0.9933 | 4.3390 | H7 | 3.3350 | 2.0316 | 2.5490 | 3.7055 | 4.4256 | 0.9933 | | 1.7226 | 5.2325 | H8 | 2.5228 | 2.0556 | 3.1767 | 2.3448 | 3.8464 | 0.9933 | 1.7226 | | 4.4238 | H9 | 1.9399 | 3.2788 | 3.6992 | 2.4194 | 0.9568 | 4.3390 | 5.2325 | 4.4238 | |
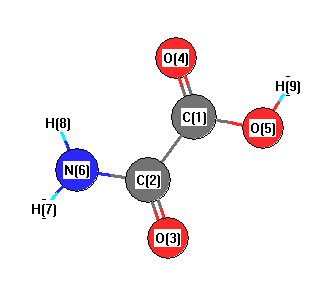 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
122.204 |
|
C1 |
C2 |
N6 |
112.394 |
| C1 |
O5 |
H9 |
114.134 |
|
C2 |
C1 |
O4 |
122.013 |
| C2 |
C1 |
O5 |
114.016 |
|
C2 |
N6 |
H7 |
118.665 |
| C2 |
N6 |
H8 |
121.081 |
|
O3 |
C2 |
N6 |
125.403 |
| O4 |
C1 |
O5 |
123.971 |
|
H7 |
N6 |
H8 |
120.253 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/CEP-121G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.205 |
|
|
|
| 2 |
C |
0.238 |
|
|
|
| 3 |
O |
-0.257 |
|
|
|
| 4 |
O |
-0.290 |
|
|
|
| 5 |
O |
-0.440 |
|
|
|
| 6 |
N |
-0.656 |
|
|
|
| 7 |
H |
0.373 |
|
|
|
| 8 |
H |
0.397 |
|
|
|
| 9 |
H |
0.429 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.576 |
0.916 |
0.000 |
2.734 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.557 |
-8.001 |
-0.003 |
| y |
-8.001 |
-44.219 |
0.004 |
| z |
-0.003 |
0.004 |
-34.188 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
15.647 |
-8.001 |
-0.003 |
| y |
-8.001 |
-15.347 |
0.004 |
| z |
-0.003 |
0.004 |
-0.300 |
|
| Polar |
|---|
| 3z2-r2 | -0.601 |
|---|
| x2-y2 | 20.662 |
|---|
| xy | -8.001 |
|---|
| xz | -0.003 |
|---|
| yz | 0.004 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.113 |
-0.028 |
0.000 |
| y |
-0.028 |
6.672 |
0.000 |
| z |
0.000 |
0.000 |
2.785 |
<r2> (average value of r
2) Å
2
| <r2> |
116.955 |
| (<r2>)1/2 |
10.815 |
Jump to
S1C1
Energy calculated at HF/CEP-121G
| | hartrees |
|---|
| Energy at 0K | -69.351823 |
| Energy at 298.15K | -69.357449 |
| Nuclear repulsion energy | 125.542355 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/CEP-121G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3956 |
3610 |
172.18 |
|
|
|
| 2 |
A' |
3955 |
3609 |
72.42 |
|
|
|
| 3 |
A' |
3798 |
3466 |
95.46 |
|
|
|
| 4 |
A' |
1928 |
1760 |
182.33 |
|
|
|
| 5 |
A' |
1832 |
1672 |
674.22 |
|
|
|
| 6 |
A' |
1794 |
1637 |
55.15 |
|
|
|
| 7 |
A' |
1547 |
1411 |
16.16 |
|
|
|
| 8 |
A' |
1361 |
1242 |
682.49 |
|
|
|
| 9 |
A' |
1285 |
1172 |
7.18 |
|
|
|
| 10 |
A' |
1215 |
1109 |
2.22 |
|
|
|
| 11 |
A' |
851 |
777 |
15.47 |
|
|
|
| 12 |
A' |
659 |
601 |
24.72 |
|
|
|
| 13 |
A' |
587 |
535 |
3.42 |
|
|
|
| 14 |
A' |
431 |
394 |
6.27 |
|
|
|
| 15 |
A' |
283 |
259 |
44.23 |
|
|
|
| 16 |
A" |
880 |
803 |
9.63 |
|
|
|
| 17 |
A" |
738 |
673 |
539.06 |
|
|
|
| 18 |
A" |
676 |
617 |
168.34 |
|
|
|
| 19 |
A" |
659 |
601 |
0.00 |
|
|
|
| 20 |
A" |
474 |
432 |
8.06 |
|
|
|
| 21 |
A" |
102 |
93 |
14.02 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14504.5 cm
-1
Scaled (by 0.9125) Zero Point Vibrational Energy (zpe) 13235.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/CEP-121G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.017 |
-0.788 |
0.000 |
| C2 |
0.000 |
0.752 |
0.000 |
| O3 |
-1.095 |
1.329 |
0.000 |
| O4 |
1.052 |
-1.422 |
0.000 |
| O5 |
-1.205 |
-1.346 |
0.000 |
| N6 |
1.213 |
1.335 |
0.000 |
| H7 |
1.278 |
2.326 |
0.000 |
| H8 |
2.038 |
0.780 |
0.000 |
| H9 |
-1.928 |
-0.717 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5404 | 2.3914 | 1.2139 | 1.3430 | 2.4367 | 3.3603 | 2.5577 | 1.9464 |
C2 | 1.5404 | | 1.2375 | 2.4153 | 2.4188 | 1.3456 | 2.0278 | 2.0380 | 2.4235 | O3 | 2.3914 | 1.2375 | | 3.4897 | 2.6766 | 2.3076 | 2.5738 | 3.1803 | 2.2086 | O4 | 1.2139 | 2.4153 | 3.4897 | | 2.2585 | 2.7615 | 3.7554 | 2.4122 | 3.0627 | O5 | 1.3430 | 2.4188 | 2.6766 | 2.2585 | | 3.6096 | 4.4325 | 3.8770 | 0.9583 | N6 | 2.4367 | 1.3456 | 2.3076 | 2.7615 | 3.6096 | | 0.9939 | 0.9943 | 3.7514 | H7 | 3.3603 | 2.0278 | 2.5738 | 3.7554 | 4.4325 | 0.9939 | | 1.7234 | 4.4201 | H8 | 2.5577 | 2.0380 | 3.1803 | 2.4122 | 3.8770 | 0.9943 | 1.7234 | | 4.2386 | H9 | 1.9464 | 2.4235 | 2.2086 | 3.0627 | 0.9583 | 3.7514 | 4.4201 | 4.2386 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
118.422 |
|
C1 |
C2 |
N6 |
115.032 |
| C1 |
O5 |
H9 |
114.474 |
|
C2 |
C1 |
O4 |
122.108 |
| C2 |
C1 |
O5 |
113.876 |
|
C2 |
N6 |
H7 |
119.413 |
| C2 |
N6 |
H8 |
120.398 |
|
O3 |
C2 |
N6 |
126.547 |
| O4 |
C1 |
O5 |
124.016 |
|
H7 |
N6 |
H8 |
120.188 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/CEP-121G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.265 |
|
|
|
| 2 |
C |
0.187 |
|
|
|
| 3 |
O |
-0.342 |
|
|
|
| 4 |
O |
-0.253 |
|
|
|
| 5 |
O |
-0.481 |
|
|
|
| 6 |
N |
-0.611 |
|
|
|
| 7 |
H |
0.377 |
|
|
|
| 8 |
H |
0.402 |
|
|
|
| 9 |
H |
0.455 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.522 |
2.970 |
0.000 |
3.337 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.852 |
8.938 |
0.000 |
| y |
8.938 |
-41.094 |
0.000 |
| z |
0.000 |
0.000 |
-34.133 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.761 |
8.938 |
0.000 |
| y |
8.938 |
-8.101 |
0.000 |
| z |
0.000 |
0.000 |
2.340 |
|
| Polar |
|---|
| 3z2-r2 | 4.680 |
|---|
| x2-y2 | 9.242 |
|---|
| xy | 8.938 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.862 |
-0.512 |
0.000 |
| y |
-0.512 |
4.785 |
0.000 |
| z |
0.000 |
0.000 |
2.753 |
<r2> (average value of r
2) Å
2
| <r2> |
115.478 |
| (<r2>)1/2 |
10.746 |