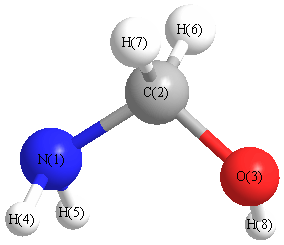
Vibrational Frequencies calculated at HF/LANL2DZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
4057 |
3651 |
19.02 |
|
|
|
| 2 |
A |
3970 |
3572 |
17.49 |
|
|
|
| 3 |
A |
3833 |
3449 |
8.45 |
|
|
|
| 4 |
A |
3369 |
3032 |
48.05 |
|
|
|
| 5 |
A |
3269 |
2942 |
91.56 |
|
|
|
| 6 |
A |
1834 |
1650 |
58.53 |
|
|
|
| 7 |
A |
1657 |
1491 |
0.71 |
|
|
|
| 8 |
A |
1557 |
1401 |
37.66 |
|
|
|
| 9 |
A |
1488 |
1339 |
1.77 |
|
|
|
| 10 |
A |
1435 |
1291 |
23.81 |
|
|
|
| 11 |
A |
1249 |
1124 |
113.47 |
|
|
|
| 12 |
A |
1173 |
1056 |
32.11 |
|
|
|
| 13 |
A |
1053 |
948 |
270.10 |
|
|
|
| 14 |
A |
968 |
871 |
2.74 |
|
|
|
| 15 |
A |
608 |
547 |
244.07 |
|
|
|
| 16 |
A |
456 |
410 |
160.80 |
|
|
|
| 17 |
A |
382 |
344 |
104.24 |
|
|
|
| 18 |
A |
253 |
228 |
223.06 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16305.8 cm
-1
Scaled (by 0.8999) Zero Point Vibrational Energy (zpe) 14673.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/LANL2DZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.759 |
|
|
|
| 2 |
C |
-0.008 |
|
|
|
| 3 |
O |
-0.631 |
|
|
|
| 4 |
H |
0.333 |
|
|
|
| 5 |
H |
0.321 |
|
|
|
| 6 |
H |
0.170 |
|
|
|
| 7 |
H |
0.199 |
|
|
|
| 8 |
H |
0.375 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.403 |
-1.083 |
1.616 |
2.398 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.531 |
-1.444 |
-2.141 |
| y |
-1.444 |
-17.087 |
-1.396 |
| z |
-2.141 |
-1.396 |
-16.562 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.707 |
-1.444 |
-2.141 |
| y |
-1.444 |
2.959 |
-1.396 |
| z |
-2.141 |
-1.396 |
3.747 |
|
| Polar |
|---|
| 3z2-r2 | 7.494 |
|---|
| x2-y2 | -6.444 |
|---|
| xy | -1.444 |
|---|
| xz | -2.141 |
|---|
| yz | -1.396 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.972 |
-0.090 |
0.008 |
| y |
-0.090 |
2.728 |
-0.198 |
| z |
0.008 |
-0.198 |
2.890 |
<r2> (average value of r
2) Å
2
| <r2> |
50.551 |
| (<r2>)1/2 |
7.110 |