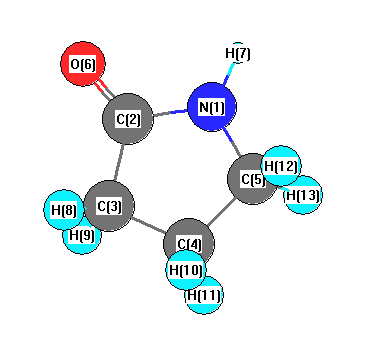
Vibrational Frequencies calculated at HF/LANL2DZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3910 |
3518 |
58.23 |
|
|
|
| 2 |
A |
3328 |
2995 |
40.27 |
|
|
|
| 3 |
A |
3311 |
2979 |
45.49 |
|
|
|
| 4 |
A |
3292 |
2962 |
42.51 |
|
|
|
| 5 |
A |
3250 |
2925 |
20.81 |
|
|
|
| 6 |
A |
3244 |
2919 |
37.98 |
|
|
|
| 7 |
A |
3225 |
2902 |
83.93 |
|
|
|
| 8 |
A |
1857 |
1671 |
616.55 |
|
|
|
| 9 |
A |
1688 |
1519 |
2.55 |
|
|
|
| 10 |
A |
1658 |
1492 |
9.93 |
|
|
|
| 11 |
A |
1631 |
1468 |
10.50 |
|
|
|
| 12 |
A |
1590 |
1431 |
33.68 |
|
|
|
| 13 |
A |
1519 |
1367 |
8.38 |
|
|
|
| 14 |
A |
1482 |
1334 |
6.15 |
|
|
|
| 15 |
A |
1452 |
1307 |
10.13 |
|
|
|
| 16 |
A |
1418 |
1276 |
189.68 |
|
|
|
| 17 |
A |
1368 |
1231 |
12.83 |
|
|
|
| 18 |
A |
1343 |
1209 |
1.33 |
|
|
|
| 19 |
A |
1308 |
1177 |
7.90 |
|
|
|
| 20 |
A |
1224 |
1101 |
2.13 |
|
|
|
| 21 |
A |
1177 |
1059 |
14.87 |
|
|
|
| 22 |
A |
1087 |
978 |
24.74 |
|
|
|
| 23 |
A |
1005 |
905 |
5.38 |
|
|
|
| 24 |
A |
992 |
893 |
4.62 |
|
|
|
| 25 |
A |
953 |
858 |
4.20 |
|
|
|
| 26 |
A |
897 |
807 |
7.14 |
|
|
|
| 27 |
A |
745 |
670 |
9.40 |
|
|
|
| 28 |
A |
724 |
651 |
124.56 |
|
|
|
| 29 |
A |
658 |
592 |
74.41 |
|
|
|
| 30 |
A |
565 |
508 |
35.78 |
|
|
|
| 31 |
A |
497 |
447 |
17.69 |
|
|
|
| 32 |
A |
205 |
184 |
0.95 |
|
|
|
| 33 |
A |
153 |
138 |
2.02 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 26376.1 cm
-1
Scaled (by 0.8999) Zero Point Vibrational Energy (zpe) 23735.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/LANL2DZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.635 |
|
|
|
| 2 |
C |
0.436 |
|
|
|
| 3 |
C |
-0.429 |
|
|
|
| 4 |
C |
-0.351 |
|
|
|
| 5 |
C |
-0.180 |
|
|
|
| 6 |
O |
-0.436 |
|
|
|
| 7 |
H |
0.388 |
|
|
|
| 8 |
H |
0.214 |
|
|
|
| 9 |
H |
0.227 |
|
|
|
| 10 |
H |
0.193 |
|
|
|
| 11 |
H |
0.196 |
|
|
|
| 12 |
H |
0.182 |
|
|
|
| 13 |
H |
0.195 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-5.146 |
-0.392 |
0.325 |
5.171 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.131 |
-0.151 |
0.302 |
| y |
-0.151 |
-31.545 |
0.107 |
| z |
0.302 |
0.107 |
-36.062 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-13.328 |
-0.151 |
0.302 |
| y |
-0.151 |
10.051 |
0.107 |
| z |
0.302 |
0.107 |
3.277 |
|
| Polar |
|---|
| 3z2-r2 | 6.553 |
|---|
| x2-y2 | -15.586 |
|---|
| xy | -0.151 |
|---|
| xz | 0.302 |
|---|
| yz | 0.107 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.239 |
-0.179 |
0.562 |
| y |
-0.179 |
6.463 |
0.043 |
| z |
0.562 |
0.043 |
5.360 |
<r2> (average value of r
2) Å
2
| <r2> |
142.208 |
| (<r2>)1/2 |
11.925 |