Jump to
S1C2
Energy calculated at HF/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -274.164620 |
| Energy at 298.15K | |
| HF Energy | -274.164620 |
| Nuclear repulsion energy | 118.888604 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2233 |
2033 |
84.96 |
35.71 |
0.06 |
0.10 |
| 2 |
A1 |
794 |
723 |
24.30 |
6.48 |
0.05 |
0.10 |
| 3 |
A1 |
605 |
551 |
131.88 |
0.79 |
0.11 |
0.20 |
| 4 |
A1 |
109 |
99 |
18.58 |
0.84 |
0.65 |
0.78 |
| 5 |
A2 |
521 |
474 |
0.00 |
1.36 |
0.75 |
0.86 |
| 6 |
B1 |
565 |
515 |
183.87 |
0.09 |
0.75 |
0.86 |
| 7 |
B2 |
2205 |
2008 |
1577.73 |
0.92 |
0.75 |
0.86 |
| 8 |
B2 |
1293 |
1177 |
80.02 |
0.27 |
0.75 |
0.86 |
| 9 |
B2 |
510 |
464 |
21.82 |
0.86 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 4417.1 cm
-1
Scaled (by 0.9106) Zero Point Vibrational Energy (zpe) 4022.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/aug-cc-pVDZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.537 |
| B2 |
0.000 |
1.236 |
0.073 |
| B3 |
0.000 |
-1.236 |
0.073 |
| O4 |
0.000 |
2.360 |
-0.314 |
| O5 |
0.000 |
-2.360 |
-0.314 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3205 | 1.3205 | 2.5092 | 2.5092 |
B2 | 1.3205 | | 2.4724 | 1.1890 | 3.6174 | B3 | 1.3205 | 2.4724 | | 3.6174 | 1.1890 | O4 | 2.5092 | 1.1890 | 3.6174 | | 4.7210 | O5 | 2.5092 | 3.6174 | 1.1890 | 4.7210 | |
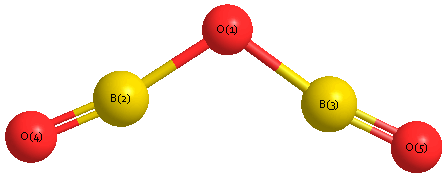 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
178.408 |
|
O1 |
B3 |
O5 |
178.408 |
| B2 |
O1 |
B3 |
138.844 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.196 |
|
|
|
| 2 |
B |
0.221 |
|
|
|
| 3 |
B |
0.221 |
|
|
|
| 4 |
O |
-0.123 |
|
|
|
| 5 |
O |
-0.123 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.578 |
1.578 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.359 |
0.000 |
0.000 |
| y |
0.000 |
-45.683 |
0.000 |
| z |
0.000 |
0.000 |
-25.445 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
11.205 |
0.000 |
0.000 |
| y |
0.000 |
-20.780 |
0.000 |
| z |
0.000 |
0.000 |
9.575 |
|
| Polar |
|---|
| 3z2-r2 | 19.151 |
|---|
| x2-y2 | 21.324 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.241 |
0.000 |
0.000 |
| y |
0.000 |
5.907 |
0.000 |
| z |
0.000 |
0.000 |
3.449 |
<r2> (average value of r
2) Å
2
| <r2> |
128.249 |
| (<r2>)1/2 |
11.325 |
Jump to
S1C1
Energy calculated at HF/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -274.162142 |
| Energy at 298.15K | |
| HF Energy | -274.162142 |
| Nuclear repulsion energy | 118.086897 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2233 |
2034 |
0.00 |
45.11 |
0.07 |
0.14 |
| 2 |
Σg |
702 |
639 |
0.00 |
6.13 |
0.07 |
0.14 |
| 3 |
Σu |
2218 |
2020 |
2082.16 |
0.00 |
0.00 |
0.00 |
| 4 |
Σu |
1370 |
1247 |
89.02 |
0.00 |
0.00 |
0.00 |
| 5 |
Πg |
532 |
484 |
0.00 |
1.47 |
0.75 |
0.86 |
| 5 |
Πg |
532 |
484 |
0.00 |
1.47 |
0.75 |
0.86 |
| 6 |
Πu |
541 |
493 |
196.08 |
0.00 |
0.00 |
0.00 |
| 6 |
Πu |
541 |
493 |
196.08 |
0.00 |
0.00 |
0.00 |
| 7 |
Πu |
92i |
84i |
8.15 |
0.00 |
0.00 |
0.00 |
| 7 |
Πu |
92i |
84i |
8.15 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 4242.2 cm
-1
Scaled (by 0.9106) Zero Point Vibrational Energy (zpe) 3862.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/aug-cc-pVDZ
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.000 |
| B2 |
0.000 |
0.000 |
1.307 |
| B3 |
0.000 |
0.000 |
-1.307 |
| O4 |
0.000 |
0.000 |
2.497 |
| O5 |
0.000 |
0.000 |
-2.497 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3065 | 1.3065 | 2.4965 | 2.4965 |
B2 | 1.3065 | | 2.6131 | 1.1900 | 3.8030 | B3 | 1.3065 | 2.6131 | | 3.8030 | 1.1900 | O4 | 2.4965 | 1.1900 | 3.8030 | | 4.9930 | O5 | 2.4965 | 3.8030 | 1.1900 | 4.9930 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
180.000 |
|
O1 |
B3 |
O5 |
180.000 |
| B2 |
O1 |
B3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
0.263 |
|
|
|
| 2 |
B |
0.155 |
|
|
|
| 3 |
B |
0.155 |
|
|
|
| 4 |
O |
-0.287 |
|
|
|
| 5 |
O |
-0.287 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.338 |
0.000 |
0.000 |
| y |
0.000 |
-24.338 |
0.000 |
| z |
0.000 |
0.000 |
-48.677 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
12.169 |
0.000 |
0.000 |
| y |
0.000 |
12.169 |
0.000 |
| z |
0.000 |
0.000 |
-24.339 |
|
| Polar |
|---|
| 3z2-r2 | -48.678 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.231 |
0.000 |
0.000 |
| y |
0.000 |
3.231 |
0.000 |
| z |
0.000 |
0.000 |
6.361 |
<r2> (average value of r
2) Å
2
| <r2> |
137.059 |
| (<r2>)1/2 |
11.707 |