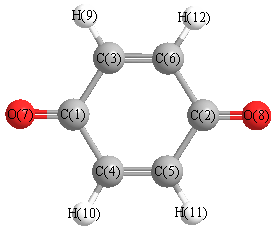
Vibrational Frequencies calculated at HF/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3734 |
3049 |
0.00 |
|
|
|
| 2 |
Ag |
2073 |
1693 |
0.00 |
|
|
|
| 3 |
Ag |
1989 |
1624 |
0.00 |
|
|
|
| 4 |
Ag |
1351 |
1103 |
0.00 |
|
|
|
| 5 |
Ag |
854 |
698 |
0.00 |
|
|
|
| 6 |
Ag |
490 |
400 |
0.00 |
|
|
|
| 7 |
Au |
1222 |
998 |
0.00 |
|
|
|
| 8 |
Au |
426 |
348 |
0.00 |
|
|
|
| 9 |
B1g |
878 |
717 |
0.00 |
|
|
|
| 10 |
B1u |
3706 |
3026 |
64.01 |
|
|
|
| 11 |
B1u |
2055 |
1678 |
42.61 |
|
|
|
| 12 |
B1u |
1614 |
1318 |
4.41 |
|
|
|
| 13 |
B1u |
1053 |
860 |
1.40 |
|
|
|
| 14 |
B1u |
850 |
694 |
0.01 |
|
|
|
| 15 |
B2g |
1221 |
997 |
0.00 |
|
|
|
| 16 |
B2g |
883 |
721 |
0.00 |
|
|
|
| 17 |
B2g |
271 |
221 |
0.00 |
|
|
|
| 18 |
B2u |
3732 |
3047 |
5.56 |
|
|
|
| 19 |
B2u |
2000 |
1633 |
4.61 |
|
|
|
| 20 |
B2u |
1487 |
1214 |
82.22 |
|
|
|
| 21 |
B2u |
1251 |
1022 |
19.88 |
|
|
|
| 22 |
B2u |
435 |
355 |
20.95 |
|
|
|
| 23 |
B3g |
3705 |
3025 |
0.00 |
|
|
|
| 24 |
B3g |
1632 |
1333 |
0.00 |
|
|
|
| 25 |
B3g |
1395 |
1139 |
0.00 |
|
|
|
| 26 |
B3g |
679 |
554 |
0.00 |
|
|
|
| 27 |
B3g |
504 |
411 |
0.00 |
|
|
|
| 28 |
B3u |
1006 |
822 |
37.13 |
|
|
|
| 29 |
B3u |
545 |
445 |
0.20 |
|
|
|
| 30 |
B3u |
86 |
70 |
5.84 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21563.8 cm
-1
Scaled (by 0.8165) Zero Point Vibrational Energy (zpe) 17606.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.186 |
|
|
|
| 2 |
C |
0.186 |
|
|
|
| 3 |
C |
-0.073 |
|
|
|
| 4 |
C |
-0.073 |
|
|
|
| 5 |
C |
-0.073 |
|
|
|
| 6 |
C |
-0.073 |
|
|
|
| 7 |
O |
-0.202 |
|
|
|
| 8 |
O |
-0.202 |
|
|
|
| 9 |
H |
0.081 |
|
|
|
| 10 |
H |
0.081 |
|
|
|
| 11 |
H |
0.081 |
|
|
|
| 12 |
H |
0.081 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.657 |
0.000 |
0.000 |
| y |
0.000 |
-37.405 |
0.000 |
| z |
0.000 |
0.000 |
-53.366 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.728 |
0.000 |
0.000 |
| y |
0.000 |
9.607 |
0.000 |
| z |
0.000 |
0.000 |
-14.335 |
|
| Polar |
|---|
| 3z2-r2 | -28.671 |
|---|
| x2-y2 | -3.252 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.250 |
0.000 |
0.000 |
| y |
0.000 |
4.869 |
0.000 |
| z |
0.000 |
0.000 |
11.253 |
<r2> (average value of r
2) Å
2
| <r2> |
244.050 |
| (<r2>)1/2 |
15.622 |