Jump to
S1C2
Energy calculated at HF/STO-3G
| | hartrees |
|---|
| Energy at 0K | -264.555506 |
| Energy at 298.15K | -264.565208 |
| Nuclear repulsion energy | 197.053784 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3722 |
3039 |
0.00 |
|
|
|
| 2 |
A |
3692 |
3015 |
5.88 |
|
|
|
| 3 |
A |
3530 |
2882 |
12.04 |
|
|
|
| 4 |
A |
3524 |
2878 |
1.51 |
|
|
|
| 5 |
A |
1825 |
1490 |
2.86 |
|
|
|
| 6 |
A |
1803 |
1472 |
2.60 |
|
|
|
| 7 |
A |
1790 |
1462 |
1.28 |
|
|
|
| 8 |
A |
1750 |
1429 |
8.83 |
|
|
|
| 9 |
A |
1525 |
1245 |
0.18 |
|
|
|
| 10 |
A |
1399 |
1142 |
23.56 |
|
|
|
| 11 |
A |
1340 |
1094 |
1.87 |
|
|
|
| 12 |
A |
1315 |
1073 |
7.21 |
|
|
|
| 13 |
A |
1087 |
887 |
6.59 |
|
|
|
| 14 |
A |
652 |
533 |
8.39 |
|
|
|
| 15 |
A |
358 |
292 |
5.87 |
|
|
|
| 16 |
A |
182 |
149 |
0.00 |
|
|
|
| 17 |
A |
111 |
91 |
2.61 |
|
|
|
| 18 |
B |
3722 |
3039 |
1.65 |
|
|
|
| 19 |
B |
3692 |
3014 |
4.41 |
|
|
|
| 20 |
B |
3648 |
2979 |
3.65 |
|
|
|
| 21 |
B |
3524 |
2877 |
10.94 |
|
|
|
| 22 |
B |
1822 |
1488 |
4.78 |
|
|
|
| 23 |
B |
1803 |
1472 |
1.34 |
|
|
|
| 24 |
B |
1761 |
1438 |
0.44 |
|
|
|
| 25 |
B |
1650 |
1348 |
111.26 |
|
|
|
| 26 |
B |
1445 |
1180 |
37.25 |
|
|
|
| 27 |
B |
1436 |
1172 |
27.72 |
|
|
|
| 28 |
B |
1313 |
1072 |
3.02 |
|
|
|
| 29 |
B |
1229 |
1004 |
2.59 |
|
|
|
| 30 |
B |
1132 |
924 |
7.84 |
|
|
|
| 31 |
B |
520 |
425 |
2.53 |
|
|
|
| 32 |
B |
230 |
188 |
9.30 |
|
|
|
| 33 |
B |
153 |
125 |
4.42 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29342.6 cm
-1
Scaled (by 0.8165) Zero Point Vibrational Energy (zpe) 23958.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/STO-3G
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.948 |
| H2 |
-0.692 |
0.569 |
1.588 |
| H3 |
0.692 |
-0.569 |
1.588 |
| O4 |
0.819 |
0.868 |
0.159 |
| O5 |
-0.819 |
-0.868 |
0.159 |
| C6 |
0.000 |
1.797 |
-0.567 |
| C7 |
0.000 |
-1.797 |
-0.567 |
| H8 |
0.667 |
2.410 |
-1.177 |
| H9 |
-0.667 |
-2.410 |
-1.177 |
| H10 |
-0.713 |
1.293 |
-1.226 |
| H11 |
-0.560 |
2.454 |
0.106 |
| H12 |
0.713 |
-1.293 |
-1.226 |
| H13 |
0.560 |
-2.454 |
0.106 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
H3 |
O4 |
O5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.1014 | 1.1014 | 1.4309 | 1.4309 | 2.3506 | 2.3506 | 3.2808 | 3.2808 | 2.6273 | 2.6542 | 2.6273 | 2.6542 |
H2 | 1.1014 | | 1.7917 | 2.1014 | 2.0316 | 2.5754 | 3.2750 | 3.5885 | 4.0645 | 2.9056 | 2.4015 | 3.6549 | 3.5923 | H3 | 1.1014 | 1.7917 | | 2.0316 | 2.1014 | 3.2750 | 2.5754 | 4.0645 | 3.5885 | 3.6549 | 3.5923 | 2.9056 | 2.4015 | O4 | 1.4309 | 2.1014 | 2.0316 | | 2.3872 | 1.4354 | 2.8816 | 2.0449 | 3.8387 | 2.1076 | 2.1022 | 2.5687 | 3.3330 | O5 | 1.4309 | 2.0316 | 2.1014 | 2.3872 | | 2.8816 | 1.4354 | 3.8387 | 2.0449 | 2.5687 | 3.3330 | 2.1076 | 2.1022 | C6 | 2.3506 | 2.5754 | 3.2750 | 1.4354 | 2.8816 | | 3.5948 | 1.0913 | 4.3030 | 1.0937 | 1.0946 | 3.2390 | 4.3408 | C7 | 2.3506 | 3.2750 | 2.5754 | 2.8816 | 1.4354 | 3.5948 | | 4.3030 | 1.0913 | 3.2390 | 4.3408 | 1.0937 | 1.0946 | H8 | 3.2808 | 3.5885 | 4.0645 | 2.0449 | 3.8387 | 1.0913 | 4.3030 | | 5.0006 | 1.7756 | 1.7754 | 3.7032 | 5.0313 | H9 | 3.2808 | 4.0645 | 3.5885 | 3.8387 | 2.0449 | 4.3030 | 1.0913 | 5.0006 | | 3.7032 | 5.0313 | 1.7756 | 1.7754 | H10 | 2.6273 | 2.9056 | 3.6549 | 2.1076 | 2.5687 | 1.0937 | 3.2390 | 1.7756 | 3.7032 | | 1.7736 | 2.9525 | 4.1753 | H11 | 2.6542 | 2.4015 | 3.5923 | 2.1022 | 3.3330 | 1.0946 | 4.3408 | 1.7754 | 5.0313 | 1.7736 | | 4.1753 | 5.0345 | H12 | 2.6273 | 3.6549 | 2.9056 | 2.5687 | 2.1076 | 3.2390 | 1.0937 | 3.7032 | 1.7756 | 2.9525 | 4.1753 | | 1.7736 | H13 | 2.6542 | 3.5923 | 2.4015 | 3.3330 | 2.1022 | 4.3408 | 1.0946 | 5.0313 | 1.7754 | 4.1753 | 5.0345 | 1.7736 | |
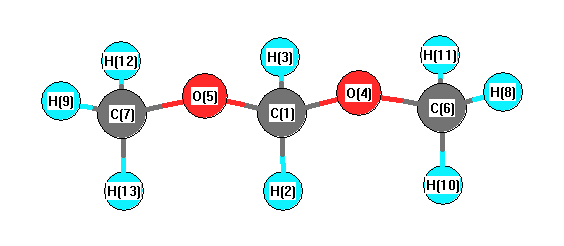 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O4 |
C6 |
110.185 |
|
C1 |
O5 |
C7 |
110.185 |
| H2 |
C1 |
H3 |
108.858 |
|
H2 |
C1 |
O4 |
111.505 |
| H2 |
C1 |
O5 |
105.964 |
|
H3 |
C1 |
O4 |
105.964 |
| H3 |
C1 |
O5 |
111.505 |
|
O4 |
C1 |
O5 |
113.062 |
| O4 |
C6 |
H8 |
107.277 |
|
O4 |
C6 |
H10 |
112.185 |
| O4 |
C6 |
H11 |
111.674 |
|
O5 |
C7 |
H9 |
107.277 |
| O5 |
C7 |
H12 |
112.185 |
|
O5 |
C7 |
H13 |
111.674 |
| H8 |
C6 |
H10 |
108.706 |
|
H8 |
C6 |
H11 |
108.623 |
| H9 |
C7 |
H12 |
108.706 |
|
H9 |
C7 |
H13 |
108.623 |
| H10 |
C6 |
H11 |
108.281 |
|
H12 |
C7 |
H13 |
108.281 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.120 |
|
|
|
| 2 |
H |
0.063 |
|
|
|
| 3 |
H |
0.063 |
|
|
|
| 4 |
O |
-0.251 |
|
|
|
| 5 |
O |
-0.251 |
|
|
|
| 6 |
C |
-0.069 |
|
|
|
| 7 |
C |
-0.069 |
|
|
|
| 8 |
H |
0.071 |
|
|
|
| 9 |
H |
0.071 |
|
|
|
| 10 |
H |
0.067 |
|
|
|
| 11 |
H |
0.058 |
|
|
|
| 12 |
H |
0.067 |
|
|
|
| 13 |
H |
0.058 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.050 |
0.050 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.268 |
-2.417 |
0.000 |
| y |
-2.417 |
-26.996 |
0.000 |
| z |
0.000 |
0.000 |
-27.712 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-4.914 |
-2.417 |
0.000 |
| y |
-2.417 |
2.993 |
0.000 |
| z |
0.000 |
0.000 |
1.920 |
|
| Polar |
|---|
| 3z2-r2 | 3.841 |
|---|
| x2-y2 | -5.272 |
|---|
| xy | -2.417 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.539 |
-0.021 |
0.000 |
| y |
-0.021 |
3.803 |
0.000 |
| z |
0.000 |
0.000 |
3.116 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |
Jump to
S1C1
Energy calculated at HF/STO-3G
| | hartrees |
|---|
| Energy at 0K | -264.548930 |
| Energy at 298.15K | -264.558460 |
| Nuclear repulsion energy | 190.806494 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3721 |
3038 |
1.66 |
|
|
|
| 2 |
A1 |
3518 |
2872 |
3.98 |
|
|
|
| 3 |
A1 |
3472 |
2835 |
10.23 |
|
|
|
| 4 |
A1 |
1846 |
1507 |
0.64 |
|
|
|
| 5 |
A1 |
1824 |
1489 |
0.77 |
|
|
|
| 6 |
A1 |
1759 |
1436 |
0.57 |
|
|
|
| 7 |
A1 |
1473 |
1203 |
2.38 |
|
|
|
| 8 |
A1 |
1387 |
1133 |
2.47 |
|
|
|
| 9 |
A1 |
1170 |
955 |
6.85 |
|
|
|
| 10 |
A1 |
514 |
420 |
0.00 |
|
|
|
| 11 |
A1 |
244 |
199 |
0.67 |
|
|
|
| 12 |
A2 |
3681 |
3006 |
0.00 |
|
|
|
| 13 |
A2 |
1806 |
1474 |
0.00 |
|
|
|
| 14 |
A2 |
1424 |
1163 |
0.00 |
|
|
|
| 15 |
A2 |
1314 |
1073 |
0.00 |
|
|
|
| 16 |
A2 |
247 |
202 |
0.00 |
|
|
|
| 17 |
A2 |
110 |
90 |
0.00 |
|
|
|
| 18 |
B1 |
3681 |
3006 |
15.32 |
|
|
|
| 19 |
B1 |
3581 |
2924 |
25.81 |
|
|
|
| 20 |
B1 |
1806 |
1474 |
6.88 |
|
|
|
| 21 |
B1 |
1320 |
1078 |
19.83 |
|
|
|
| 22 |
B1 |
1240 |
1013 |
16.66 |
|
|
|
| 23 |
B1 |
247 |
202 |
4.40 |
|
|
|
| 24 |
B1 |
101 |
83 |
0.16 |
|
|
|
| 25 |
B2 |
3721 |
3038 |
0.05 |
|
|
|
| 26 |
B2 |
3518 |
2872 |
9.57 |
|
|
|
| 27 |
B2 |
1824 |
1490 |
5.51 |
|
|
|
| 28 |
B2 |
1766 |
1442 |
1.29 |
|
|
|
| 29 |
B2 |
1690 |
1380 |
161.30 |
|
|
|
| 30 |
B2 |
1431 |
1169 |
43.61 |
|
|
|
| 31 |
B2 |
1361 |
1111 |
0.01 |
|
|
|
| 32 |
B2 |
1189 |
971 |
0.05 |
|
|
|
| 33 |
B2 |
521 |
425 |
0.77 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29253.6 cm
-1
Scaled (by 0.8165) Zero Point Vibrational Energy (zpe) 23885.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/STO-3G
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.316 |
| H2 |
-0.893 |
0.000 |
0.970 |
| H3 |
0.893 |
0.000 |
0.970 |
| O4 |
0.000 |
1.132 |
-0.559 |
| O5 |
0.000 |
-1.132 |
-0.559 |
| C6 |
0.000 |
2.335 |
0.220 |
| C7 |
0.000 |
-2.335 |
0.220 |
| H8 |
0.000 |
3.174 |
-0.478 |
| H9 |
0.000 |
-3.174 |
-0.478 |
| H10 |
-0.887 |
2.412 |
0.857 |
| H11 |
0.887 |
2.412 |
0.857 |
| H12 |
0.887 |
-2.412 |
0.857 |
| H13 |
-0.887 |
-2.412 |
0.857 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
H3 |
O4 |
O5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.1064 | 1.1064 | 1.4307 | 1.4307 | 2.3371 | 2.3371 | 3.2716 | 3.2716 | 2.6267 | 2.6267 | 2.6267 | 2.6267 |
H2 | 1.1064 | | 1.7853 | 2.1012 | 2.1012 | 2.6100 | 2.6100 | 3.6009 | 3.6009 | 2.4151 | 3.0001 | 3.0001 | 2.4151 | H3 | 1.1064 | 1.7853 | | 2.1012 | 2.1012 | 2.6100 | 2.6100 | 3.6009 | 3.6009 | 3.0001 | 2.4151 | 2.4151 | 3.0001 | O4 | 1.4307 | 2.1012 | 2.1012 | | 2.2637 | 1.4333 | 3.5533 | 2.0434 | 4.3062 | 2.1051 | 2.1051 | 3.9184 | 3.9184 | O5 | 1.4307 | 2.1012 | 2.1012 | 2.2637 | | 3.5533 | 1.4333 | 4.3062 | 2.0434 | 3.9184 | 3.9184 | 2.1051 | 2.1051 | C6 | 2.3371 | 2.6100 | 2.6100 | 1.4333 | 3.5533 | | 4.6702 | 1.0912 | 5.5528 | 1.0949 | 1.0949 | 4.8716 | 4.8716 | C7 | 2.3371 | 2.6100 | 2.6100 | 3.5533 | 1.4333 | 4.6702 | | 5.5528 | 1.0912 | 4.8716 | 4.8716 | 1.0949 | 1.0949 | H8 | 3.2716 | 3.6009 | 3.6009 | 2.0434 | 4.3062 | 1.0912 | 5.5528 | | 6.3472 | 1.7746 | 1.7746 | 5.8116 | 5.8116 | H9 | 3.2716 | 3.6009 | 3.6009 | 4.3062 | 2.0434 | 5.5528 | 1.0912 | 6.3472 | | 5.8116 | 5.8116 | 1.7746 | 1.7746 | H10 | 2.6267 | 2.4151 | 3.0001 | 2.1051 | 3.9184 | 1.0949 | 4.8716 | 1.7746 | 5.8116 | | 1.7743 | 5.1409 | 4.8250 | H11 | 2.6267 | 3.0001 | 2.4151 | 2.1051 | 3.9184 | 1.0949 | 4.8716 | 1.7746 | 5.8116 | 1.7743 | | 4.8250 | 5.1409 | H12 | 2.6267 | 3.0001 | 2.4151 | 3.9184 | 2.1051 | 4.8716 | 1.0949 | 5.8116 | 1.7746 | 5.1409 | 4.8250 | | 1.7743 | H13 | 2.6267 | 2.4151 | 3.0001 | 3.9184 | 2.1051 | 4.8716 | 1.0949 | 5.8116 | 1.7746 | 4.8250 | 5.1409 | 1.7743 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O4 |
C6 |
109.379 |
|
C1 |
O5 |
C7 |
109.379 |
| H2 |
C1 |
H3 |
107.571 |
|
H2 |
C1 |
O4 |
111.185 |
| H2 |
C1 |
O5 |
111.185 |
|
H3 |
C1 |
O4 |
111.185 |
| H3 |
C1 |
O5 |
111.185 |
|
O4 |
C1 |
O5 |
104.578 |
| O4 |
C6 |
H8 |
107.305 |
|
O4 |
C6 |
H10 |
112.054 |
| O4 |
C6 |
H11 |
112.054 |
|
O5 |
C7 |
H9 |
107.305 |
| O5 |
C7 |
H12 |
112.054 |
|
O5 |
C7 |
H13 |
112.054 |
| H8 |
C6 |
H10 |
108.541 |
|
H8 |
C6 |
H11 |
108.541 |
| H9 |
C7 |
H12 |
108.541 |
|
H9 |
C7 |
H13 |
108.541 |
| H10 |
C6 |
H11 |
108.236 |
|
H12 |
C7 |
H13 |
108.236 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.128 |
|
|
|
| 2 |
H |
0.046 |
|
|
|
| 3 |
H |
0.046 |
|
|
|
| 4 |
O |
-0.235 |
|
|
|
| 5 |
O |
-0.235 |
|
|
|
| 6 |
C |
-0.066 |
|
|
|
| 7 |
C |
-0.066 |
|
|
|
| 8 |
H |
0.075 |
|
|
|
| 9 |
H |
0.075 |
|
|
|
| 10 |
H |
0.058 |
|
|
|
| 11 |
H |
0.058 |
|
|
|
| 12 |
H |
0.058 |
|
|
|
| 13 |
H |
0.058 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
2.592 |
2.592 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.336 |
0.000 |
0.000 |
| y |
0.000 |
-25.110 |
0.000 |
| z |
0.000 |
0.000 |
-30.880 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.341 |
0.000 |
0.000 |
| y |
0.000 |
4.998 |
0.000 |
| z |
0.000 |
0.000 |
-3.656 |
|
| Polar |
|---|
| 3z2-r2 | -7.313 |
|---|
| x2-y2 | -4.226 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.425 |
0.000 |
0.000 |
| y |
0.000 |
4.752 |
0.000 |
| z |
0.000 |
0.000 |
2.565 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |