Jump to
S1C2
Energy calculated at HF/STO-3G
| | hartrees |
|---|
| Energy at 0K | -336.097121 |
| Energy at 298.15K | |
| HF Energy | -336.097121 |
| Nuclear repulsion energy | 241.689956 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3584 |
2927 |
2.37 |
|
|
|
| 2 |
A1 |
2165 |
1768 |
253.63 |
|
|
|
| 3 |
A1 |
1829 |
1493 |
3.23 |
|
|
|
| 4 |
A1 |
1617 |
1320 |
5.10 |
|
|
|
| 5 |
A1 |
1412 |
1153 |
76.30 |
|
|
|
| 6 |
A1 |
1136 |
927 |
11.63 |
|
|
|
| 7 |
A1 |
935 |
763 |
18.04 |
|
|
|
| 8 |
A1 |
764 |
624 |
0.39 |
|
|
|
| 9 |
A2 |
3690 |
3013 |
0.00 |
|
|
|
| 10 |
A2 |
1448 |
1182 |
0.00 |
|
|
|
| 11 |
A2 |
1298 |
1060 |
0.00 |
|
|
|
| 12 |
A2 |
111i |
91i |
0.00 |
|
|
|
| 13 |
B1 |
3696 |
3018 |
1.35 |
|
|
|
| 14 |
B1 |
1408 |
1149 |
7.29 |
|
|
|
| 15 |
B1 |
970 |
792 |
1.99 |
|
|
|
| 16 |
B1 |
703 |
574 |
32.06 |
|
|
|
| 17 |
B1 |
87 |
71 |
2.49 |
|
|
|
| 18 |
B2 |
3580 |
2923 |
0.69 |
|
|
|
| 19 |
B2 |
1816 |
1483 |
0.00 |
|
|
|
| 20 |
B2 |
1641 |
1340 |
5.44 |
|
|
|
| 21 |
B2 |
1458 |
1190 |
81.84 |
|
|
|
| 22 |
B2 |
1258 |
1027 |
86.70 |
|
|
|
| 23 |
B2 |
857 |
700 |
1.21 |
|
|
|
| 24 |
B2 |
520 |
424 |
4.88 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18878.7 cm
-1
Scaled (by 0.8165) Zero Point Vibrational Energy (zpe) 15414.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/STO-3G
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.864 |
| O2 |
0.000 |
0.000 |
2.070 |
| O3 |
0.000 |
1.172 |
0.065 |
| O4 |
0.000 |
-1.172 |
0.065 |
| C5 |
0.000 |
0.780 |
-1.299 |
| C6 |
0.000 |
-0.780 |
-1.299 |
| H7 |
-0.888 |
1.180 |
-1.799 |
| H8 |
0.888 |
1.180 |
-1.799 |
| H9 |
0.888 |
-1.180 |
-1.799 |
| H10 |
-0.888 |
-1.180 |
-1.799 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.2062 | 1.4190 | 1.4190 | 2.2995 | 2.2995 | 3.0453 | 3.0453 | 3.0453 | 3.0453 |
O2 | 1.2062 | | 2.3232 | 2.3232 | 3.4585 | 3.4585 | 4.1417 | 4.1417 | 4.1417 | 4.1417 | O3 | 1.4190 | 2.3232 | | 2.3447 | 1.4190 | 2.3813 | 2.0644 | 2.0644 | 3.1298 | 3.1298 | O4 | 1.4190 | 2.3232 | 2.3447 | | 2.3813 | 1.4190 | 3.1298 | 3.1298 | 2.0644 | 2.0644 | C5 | 2.2995 | 3.4585 | 1.4190 | 2.3813 | | 1.5598 | 1.0946 | 1.0946 | 2.2089 | 2.2089 | C6 | 2.2995 | 3.4585 | 2.3813 | 1.4190 | 1.5598 | | 2.2089 | 2.2089 | 1.0946 | 1.0946 | H7 | 3.0453 | 4.1417 | 2.0644 | 3.1298 | 1.0946 | 2.2089 | | 1.7751 | 2.9532 | 2.3602 | H8 | 3.0453 | 4.1417 | 2.0644 | 3.1298 | 1.0946 | 2.2089 | 1.7751 | | 2.3602 | 2.9532 | H9 | 3.0453 | 4.1417 | 3.1298 | 2.0644 | 2.2089 | 1.0946 | 2.9532 | 2.3602 | | 1.7751 | H10 | 3.0453 | 4.1417 | 3.1298 | 2.0644 | 2.2089 | 1.0946 | 2.3602 | 2.9532 | 1.7751 | |
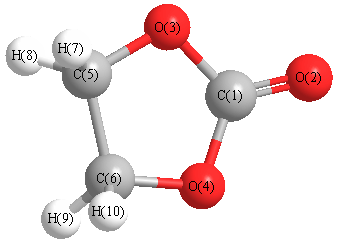 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
108.236 |
|
C1 |
O4 |
C6 |
108.236 |
| O2 |
C1 |
O3 |
124.292 |
|
O2 |
C1 |
O4 |
124.292 |
| O3 |
C1 |
O4 |
111.415 |
|
O3 |
C5 |
C6 |
106.056 |
| O3 |
C5 |
H7 |
109.757 |
|
O3 |
C5 |
H8 |
109.757 |
| O4 |
C6 |
C5 |
106.056 |
|
O4 |
C6 |
H9 |
109.757 |
| O4 |
C6 |
H10 |
109.757 |
|
C5 |
C6 |
H9 |
111.445 |
| C5 |
C6 |
H10 |
111.445 |
|
C6 |
C5 |
H7 |
111.445 |
| C6 |
C5 |
H8 |
111.445 |
|
H7 |
C5 |
H8 |
108.361 |
| H9 |
C6 |
H10 |
108.361 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.418 |
|
|
|
| 2 |
O |
-0.247 |
|
|
|
| 3 |
O |
-0.241 |
|
|
|
| 4 |
O |
-0.241 |
|
|
|
| 5 |
C |
-0.002 |
|
|
|
| 6 |
C |
-0.002 |
|
|
|
| 7 |
H |
0.078 |
|
|
|
| 8 |
H |
0.078 |
|
|
|
| 9 |
H |
0.078 |
|
|
|
| 10 |
H |
0.078 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-3.690 |
3.690 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.588 |
0.000 |
0.000 |
| y |
0.000 |
-34.513 |
0.000 |
| z |
0.000 |
0.000 |
-33.264 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.300 |
0.000 |
0.000 |
| y |
0.000 |
-3.087 |
0.000 |
| z |
0.000 |
0.000 |
-1.213 |
|
| Polar |
|---|
| 3z2-r2 | -2.426 |
|---|
| x2-y2 | 4.925 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.907 |
0.000 |
0.000 |
| y |
0.000 |
2.608 |
0.000 |
| z |
0.000 |
0.000 |
4.785 |
<r2> (average value of r
2) Å
2
| <r2> |
130.318 |
| (<r2>)1/2 |
11.416 |
Jump to
S1C1
Energy calculated at HF/STO-3G
| | hartrees |
|---|
| Energy at 0K | -336.097129 |
| Energy at 298.15K | |
| HF Energy | -336.097129 |
| Nuclear repulsion energy | 241.741845 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3691 |
3014 |
0.02 |
|
|
|
| 2 |
A |
3584 |
2926 |
2.26 |
|
|
|
| 3 |
A |
2165 |
1768 |
252.06 |
|
|
|
| 4 |
A |
1827 |
1492 |
3.30 |
|
|
|
| 5 |
A |
1618 |
1321 |
5.75 |
|
|
|
| 6 |
A |
1447 |
1182 |
1.53 |
|
|
|
| 7 |
A |
1410 |
1151 |
73.60 |
|
|
|
| 8 |
A |
1297 |
1059 |
1.40 |
|
|
|
| 9 |
A |
1134 |
926 |
10.64 |
|
|
|
| 10 |
A |
935 |
763 |
17.99 |
|
|
|
| 11 |
A |
763 |
623 |
0.38 |
|
|
|
| 12 |
A |
85i |
69i |
0.05 |
|
|
|
| 13 |
B |
3697 |
3018 |
1.33 |
|
|
|
| 14 |
B |
3580 |
2923 |
0.95 |
|
|
|
| 15 |
B |
1815 |
1482 |
0.00 |
|
|
|
| 16 |
B |
1640 |
1339 |
5.49 |
|
|
|
| 17 |
B |
1456 |
1189 |
79.84 |
|
|
|
| 18 |
B |
1407 |
1149 |
9.53 |
|
|
|
| 19 |
B |
1257 |
1026 |
85.52 |
|
|
|
| 20 |
B |
980 |
800 |
1.80 |
|
|
|
| 21 |
B |
845 |
690 |
1.21 |
|
|
|
| 22 |
B |
704 |
575 |
32.41 |
|
|
|
| 23 |
B |
520 |
424 |
4.86 |
|
|
|
| 24 |
B |
91 |
74 |
2.46 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18889.3 cm
-1
Scaled (by 0.8165) Zero Point Vibrational Energy (zpe) 15423.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/STO-3G
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.864 |
| O2 |
0.000 |
0.000 |
2.070 |
| O3 |
0.000 |
1.172 |
0.063 |
| O4 |
0.000 |
-1.172 |
0.063 |
| C5 |
0.068 |
0.776 |
-1.298 |
| C6 |
-0.068 |
-0.776 |
-1.298 |
| H7 |
-0.740 |
1.253 |
-1.860 |
| H8 |
1.024 |
1.092 |
-1.731 |
| H9 |
0.740 |
-1.253 |
-1.860 |
| H10 |
-1.024 |
-1.092 |
-1.731 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.2062 | 1.4194 | 1.4194 | 2.2978 | 2.2978 | 3.0883 | 2.9951 | 3.0883 | 2.9951 |
O2 | 1.2062 | | 2.3237 | 2.3237 | 3.4567 | 3.4567 | 4.1910 | 4.0847 | 4.1910 | 4.0847 | O3 | 1.4194 | 2.3237 | | 2.3446 | 1.4194 | 2.3782 | 2.0628 | 2.0674 | 3.1829 | 3.0648 | O4 | 1.4194 | 2.3237 | 2.3446 | | 2.3782 | 1.4194 | 3.1829 | 3.0648 | 2.0628 | 2.0674 | C5 | 2.2978 | 3.4567 | 1.4194 | 2.3782 | | 1.5590 | 1.0940 | 1.0951 | 2.2104 | 2.2069 | C6 | 2.2978 | 3.4567 | 2.3782 | 1.4194 | 1.5590 | | 2.2104 | 2.2069 | 1.0940 | 1.0951 | H7 | 3.0883 | 4.1910 | 2.0628 | 3.1829 | 1.0940 | 2.2104 | | 1.7756 | 2.9102 | 2.3654 | H8 | 2.9951 | 4.0847 | 2.0674 | 3.0648 | 1.0951 | 2.2069 | 1.7756 | | 2.3654 | 2.9932 | H9 | 3.0883 | 4.1910 | 3.1829 | 2.0628 | 2.2104 | 1.0940 | 2.9102 | 2.3654 | | 1.7756 | H10 | 2.9951 | 4.0847 | 3.0648 | 2.0674 | 2.2069 | 1.0951 | 2.3654 | 2.9932 | 1.7756 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
108.082 |
|
C1 |
O4 |
C6 |
108.082 |
| O2 |
C1 |
O3 |
124.316 |
|
O2 |
C1 |
O4 |
124.316 |
| O3 |
C1 |
O4 |
111.369 |
|
O3 |
C5 |
C6 |
105.875 |
| O3 |
C5 |
H7 |
109.639 |
|
O3 |
C5 |
H8 |
109.940 |
| O4 |
C6 |
C5 |
105.875 |
|
O4 |
C6 |
H9 |
109.639 |
| O4 |
C6 |
H10 |
109.940 |
|
C5 |
C6 |
H9 |
111.655 |
| C5 |
C6 |
H10 |
111.302 |
|
C6 |
C5 |
H7 |
111.655 |
| C6 |
C5 |
H8 |
111.302 |
|
H7 |
C5 |
H8 |
108.408 |
| H9 |
C6 |
H10 |
108.408 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.418 |
|
|
|
| 2 |
O |
-0.247 |
|
|
|
| 3 |
O |
-0.241 |
|
|
|
| 4 |
O |
-0.241 |
|
|
|
| 5 |
C |
-0.002 |
|
|
|
| 6 |
C |
-0.002 |
|
|
|
| 7 |
H |
0.080 |
|
|
|
| 8 |
H |
0.077 |
|
|
|
| 9 |
H |
0.080 |
|
|
|
| 10 |
H |
0.077 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-3.684 |
3.684 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.592 |
0.052 |
0.000 |
| y |
0.052 |
-34.512 |
0.000 |
| z |
0.000 |
0.000 |
-33.249 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.289 |
0.052 |
0.000 |
| y |
0.052 |
-3.092 |
0.000 |
| z |
0.000 |
0.000 |
-1.197 |
|
| Polar |
|---|
| 3z2-r2 | -2.393 |
|---|
| x2-y2 | 4.920 |
|---|
| xy | 0.052 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.912 |
0.024 |
0.000 |
| y |
0.024 |
2.603 |
0.000 |
| z |
0.000 |
0.000 |
4.784 |
<r2> (average value of r
2) Å
2
| <r2> |
130.203 |
| (<r2>)1/2 |
11.411 |