Jump to
S1C2
Energy calculated at HF/6-31G**
| | hartrees |
|---|
| Energy at 0K | -412.480026 |
| Energy at 298.15K | |
| HF Energy | -412.480026 |
| Nuclear repulsion energy | 25.303573 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at HF/6-31G**
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
0.000 |
0.000 |
| Li2 |
0.000 |
0.000 |
2.102 |
| Li3 |
0.000 |
0.000 |
-2.102 |
Atom - Atom Distances (Å)
| |
S1 |
Li2 |
Li3 |
| S1 | | 2.1018 | 2.1018 |
Li2 | 2.1018 | | 4.2035 | Li3 | 2.1018 | 4.2035 | |
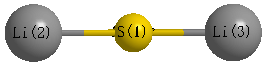 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li2 |
S1 |
Li3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
-0.621 |
|
|
|
| 2 |
Li |
0.311 |
|
|
|
| 3 |
Li |
0.311 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.642 |
0.000 |
0.000 |
| y |
0.000 |
-23.642 |
0.000 |
| z |
0.000 |
0.000 |
14.531 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-19.087 |
0.000 |
0.000 |
| y |
0.000 |
-19.087 |
0.000 |
| z |
0.000 |
0.000 |
38.173 |
|
| Polar |
|---|
| 3z2-r2 | 76.346 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.054 |
0.000 |
0.000 |
| y |
0.000 |
6.054 |
0.000 |
| z |
0.000 |
0.000 |
10.685 |
<r2> (average value of r
2) Å
2
| <r2> |
33.324 |
| (<r2>)1/2 |
5.773 |
Jump to
S1C1
Energy calculated at HF/6-31G**
| | hartrees |
|---|
| Energy at 0K | -412.480240 |
| Energy at 298.15K | -412.480478 |
| HF Energy | -412.480240 |
| Nuclear repulsion energy | 25.282626 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at HF/6-31G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
0.000 |
0.181 |
| Li2 |
0.000 |
2.002 |
-0.481 |
| Li3 |
0.000 |
-2.002 |
-0.481 |
Atom - Atom Distances (Å)
| |
S1 |
Li2 |
Li3 |
| S1 | | 2.1085 | 2.1085 |
Li2 | 2.1085 | | 4.0039 | Li3 | 2.1085 | 4.0039 | |
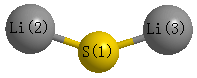 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li2 |
S1 |
Li3 |
143.407 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
-0.593 |
|
|
|
| 2 |
Li |
0.297 |
|
|
|
| 3 |
Li |
0.297 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-3.858 |
3.858 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.690 |
0.000 |
0.000 |
| y |
0.000 |
11.192 |
0.000 |
| z |
0.000 |
0.000 |
-21.581 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-18.496 |
0.000 |
0.000 |
| y |
0.000 |
33.828 |
0.000 |
| z |
0.000 |
0.000 |
-15.332 |
|
| Polar |
|---|
| 3z2-r2 | -30.663 |
|---|
| x2-y2 | -34.882 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.368 |
0.000 |
0.000 |
| y |
0.000 |
10.057 |
0.000 |
| z |
0.000 |
0.000 |
6.667 |
<r2> (average value of r
2) Å
2
| <r2> |
33.053 |
| (<r2>)1/2 |
5.749 |