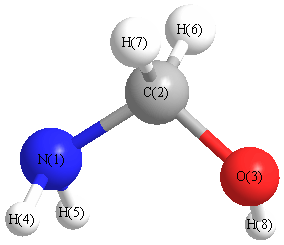
Vibrational Frequencies calculated at HF/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
4167 |
3777 |
44.68 |
|
|
|
| 2 |
A |
3817 |
3460 |
4.91 |
|
|
|
| 3 |
A |
3733 |
3384 |
2.42 |
|
|
|
| 4 |
A |
3254 |
2950 |
42.03 |
|
|
|
| 5 |
A |
3181 |
2883 |
71.35 |
|
|
|
| 6 |
A |
1801 |
1632 |
33.49 |
|
|
|
| 7 |
A |
1642 |
1488 |
0.15 |
|
|
|
| 8 |
A |
1551 |
1406 |
49.67 |
|
|
|
| 9 |
A |
1504 |
1363 |
0.03 |
|
|
|
| 10 |
A |
1486 |
1347 |
13.95 |
|
|
|
| 11 |
A |
1245 |
1129 |
51.36 |
|
|
|
| 12 |
A |
1180 |
1070 |
62.55 |
|
|
|
| 13 |
A |
1132 |
1026 |
223.33 |
|
|
|
| 14 |
A |
982 |
890 |
2.90 |
|
|
|
| 15 |
A |
891 |
808 |
148.61 |
|
|
|
| 16 |
A |
521 |
472 |
47.05 |
|
|
|
| 17 |
A |
396 |
359 |
93.08 |
|
|
|
| 18 |
A |
286 |
259 |
85.83 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16385.2 cm
-1
Scaled (by 0.9064) Zero Point Vibrational Energy (zpe) 14851.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.412 |
|
|
|
| 2 |
C |
0.277 |
|
|
|
| 3 |
O |
-0.425 |
|
|
|
| 4 |
H |
0.147 |
|
|
|
| 5 |
H |
0.130 |
|
|
|
| 6 |
H |
0.042 |
|
|
|
| 7 |
H |
0.047 |
|
|
|
| 8 |
H |
0.192 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.297 |
-1.071 |
1.301 |
1.711 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.509 |
-1.479 |
-2.122 |
| y |
-1.479 |
-17.161 |
-1.458 |
| z |
-2.122 |
-1.458 |
-16.591 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.633 |
-1.479 |
-2.122 |
| y |
-1.479 |
2.889 |
-1.458 |
| z |
-2.122 |
-1.458 |
3.744 |
|
| Polar |
|---|
| 3z2-r2 | 7.488 |
|---|
| x2-y2 | -6.348 |
|---|
| xy | -1.479 |
|---|
| xz | -2.122 |
|---|
| yz | -1.458 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.841 |
-0.046 |
-0.055 |
| y |
-0.046 |
3.629 |
-0.096 |
| z |
-0.055 |
-0.096 |
3.552 |
<r2> (average value of r
2) Å
2
| <r2> |
48.817 |
| (<r2>)1/2 |
6.987 |