Jump to
S1C2
Energy calculated at HF/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -134.309447 |
| Energy at 298.15K | -134.317756 |
| HF Energy | -134.309447 |
| Nuclear repulsion energy | 83.338844 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3720 |
3372 |
0.48 |
|
|
|
| 2 |
A' |
3207 |
2907 |
73.45 |
|
|
|
| 3 |
A' |
3168 |
2872 |
31.81 |
|
|
|
| 4 |
A' |
3145 |
2851 |
32.93 |
|
|
|
| 5 |
A' |
1794 |
1626 |
26.74 |
|
|
|
| 6 |
A' |
1627 |
1475 |
1.42 |
|
|
|
| 7 |
A' |
1605 |
1455 |
0.55 |
|
|
|
| 8 |
A' |
1535 |
1391 |
11.31 |
|
|
|
| 9 |
A' |
1501 |
1360 |
13.11 |
|
|
|
| 10 |
A' |
1232 |
1116 |
12.53 |
|
|
|
| 11 |
A' |
1143 |
1036 |
39.83 |
|
|
|
| 12 |
A' |
953 |
864 |
38.30 |
|
|
|
| 13 |
A' |
916 |
831 |
138.56 |
|
|
|
| 14 |
A' |
431 |
390 |
6.62 |
|
|
|
| 15 |
A" |
3797 |
3442 |
1.18 |
|
|
|
| 16 |
A" |
3215 |
2914 |
98.49 |
|
|
|
| 17 |
A" |
3184 |
2886 |
0.04 |
|
|
|
| 18 |
A" |
1608 |
1457 |
6.08 |
|
|
|
| 19 |
A" |
1498 |
1358 |
0.20 |
|
|
|
| 20 |
A" |
1374 |
1246 |
0.03 |
|
|
|
| 21 |
A" |
1078 |
977 |
0.78 |
|
|
|
| 22 |
A" |
834 |
756 |
0.43 |
|
|
|
| 23 |
A" |
302 |
274 |
25.80 |
|
|
|
| 24 |
A" |
264 |
239 |
22.78 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21565.1 cm
-1
Scaled (by 0.9064) Zero Point Vibrational Energy (zpe) 19546.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/Def2TZVPP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-1.297 |
-0.091 |
0.000 |
| C2 |
0.000 |
0.571 |
0.000 |
| C3 |
1.207 |
-0.351 |
0.000 |
| H4 |
2.135 |
0.211 |
0.000 |
| H5 |
1.209 |
-0.991 |
0.876 |
| H6 |
1.209 |
-0.991 |
-0.876 |
| H7 |
0.036 |
1.221 |
-0.866 |
| H8 |
0.036 |
1.221 |
0.866 |
| H9 |
-1.395 |
-0.675 |
0.804 |
| H10 |
-1.395 |
-0.675 |
-0.804 |
Atom - Atom Distances (Å)
| |
N1 |
C2 |
C3 |
H4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.4565 | 2.5176 | 3.4457 | 2.8032 | 2.8032 | 2.0611 | 2.0611 | 0.9980 | 0.9980 |
C2 | 1.4565 | | 1.5194 | 2.1656 | 2.1613 | 2.1613 | 1.0827 | 1.0827 | 2.0360 | 2.0360 | C3 | 2.5176 | 1.5194 | | 1.0854 | 1.0849 | 1.0849 | 2.1429 | 2.1429 | 2.7429 | 2.7429 | H4 | 3.4457 | 2.1656 | 1.0854 | | 1.7526 | 1.7526 | 2.4850 | 2.4850 | 3.7278 | 3.7278 | H5 | 2.8032 | 2.1613 | 1.0849 | 1.7526 | | 1.7518 | 3.0499 | 2.5039 | 2.6246 | 3.1152 | H6 | 2.8032 | 2.1613 | 1.0849 | 1.7526 | 1.7518 | | 2.5039 | 3.0499 | 3.1152 | 2.6246 | H7 | 2.0611 | 1.0827 | 2.1429 | 2.4850 | 3.0499 | 2.5039 | | 1.7311 | 2.9032 | 2.3761 | H8 | 2.0611 | 1.0827 | 2.1429 | 2.4850 | 2.5039 | 3.0499 | 1.7311 | | 2.3761 | 2.9032 | H9 | 0.9980 | 2.0360 | 2.7429 | 3.7278 | 2.6246 | 3.1152 | 2.9032 | 2.3761 | | 1.6074 | H10 | 0.9980 | 2.0360 | 2.7429 | 3.7278 | 3.1152 | 2.6246 | 2.3761 | 2.9032 | 1.6074 | |
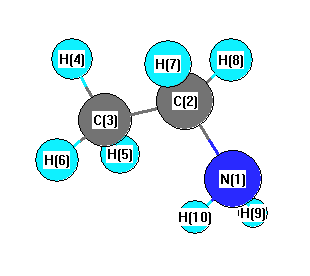 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C2 |
C3 |
115.548 |
|
N1 |
C2 |
H7 |
107.615 |
| N1 |
C2 |
H8 |
107.615 |
|
C2 |
N1 |
H9 |
110.712 |
| C2 |
N1 |
H10 |
110.712 |
|
C2 |
C3 |
H4 |
111.394 |
| C2 |
C3 |
H5 |
111.082 |
|
C2 |
C3 |
H6 |
111.082 |
| C3 |
C2 |
H7 |
109.738 |
|
C3 |
C2 |
H8 |
109.738 |
| H4 |
C3 |
H5 |
107.712 |
|
H4 |
C3 |
H6 |
107.712 |
| H5 |
C3 |
H6 |
107.685 |
|
H7 |
C2 |
H8 |
106.151 |
| H9 |
N1 |
H10 |
107.277 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.392 |
|
|
|
| 2 |
C |
0.111 |
|
|
|
| 3 |
C |
-0.163 |
|
|
|
| 4 |
H |
0.046 |
|
|
|
| 5 |
H |
0.038 |
|
|
|
| 6 |
H |
0.038 |
|
|
|
| 7 |
H |
0.034 |
|
|
|
| 8 |
H |
0.034 |
|
|
|
| 9 |
H |
0.126 |
|
|
|
| 10 |
H |
0.126 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.930 |
-0.928 |
0.000 |
1.314 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.042 |
2.650 |
0.000 |
| y |
2.650 |
-20.480 |
0.000 |
| z |
0.000 |
0.000 |
-19.102 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-4.251 |
2.650 |
0.000 |
| y |
2.650 |
1.092 |
0.000 |
| z |
0.000 |
0.000 |
3.159 |
|
| Polar |
|---|
| 3z2-r2 | 6.318 |
|---|
| x2-y2 | -3.562 |
|---|
| xy | 2.650 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.279 |
0.081 |
0.000 |
| y |
0.081 |
4.803 |
0.000 |
| z |
0.000 |
0.000 |
4.668 |
<r2> (average value of r
2) Å
2
| <r2> |
58.193 |
| (<r2>)1/2 |
7.628 |
Jump to
S1C1
Energy calculated at HF/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -134.309675 |
| Energy at 298.15K | |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/Def2TZVPP
Geometric Data calculated at HF/Def2TZVPP
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability