Jump to
S1C2
Energy calculated at HF/3-21G*
| | hartrees |
|---|
| Energy at 0K | -545.939857 |
| Energy at 298.15K | |
| HF Energy | -545.939857 |
| Nuclear repulsion energy | 352.855226 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/3-21G*
Geometric Data calculated at HF/3-21G*
Point Group is D4h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
1.010 |
0.000 |
| C2 |
1.010 |
0.000 |
0.000 |
| C3 |
0.000 |
-1.010 |
0.000 |
| C4 |
-1.010 |
0.000 |
0.000 |
| F5 |
0.000 |
2.334 |
0.000 |
| F6 |
2.334 |
0.000 |
0.000 |
| F7 |
0.000 |
-2.334 |
0.000 |
| F8 |
-2.334 |
0.000 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
| C1 | | 1.4285 | 2.0202 | 1.4285 | 1.3244 | 2.5436 | 3.3446 | 2.5436 |
C2 | 1.4285 | | 1.4285 | 2.0202 | 2.5436 | 1.3244 | 2.5436 | 3.3446 | C3 | 2.0202 | 1.4285 | | 1.4285 | 3.3446 | 2.5436 | 1.3244 | 2.5436 | C4 | 1.4285 | 2.0202 | 1.4285 | | 2.5436 | 3.3446 | 2.5436 | 1.3244 | F5 | 1.3244 | 2.5436 | 3.3446 | 2.5436 | | 3.3014 | 4.6689 | 3.3014 | F6 | 2.5436 | 1.3244 | 2.5436 | 3.3446 | 3.3014 | | 3.3014 | 4.6689 | F7 | 3.3446 | 2.5436 | 1.3244 | 2.5436 | 4.6689 | 3.3014 | | 3.3014 | F8 | 2.5436 | 3.3446 | 2.5436 | 1.3244 | 3.3014 | 4.6689 | 3.3014 | |
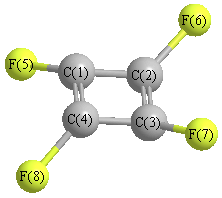 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
90.000 |
|
C1 |
C2 |
F6 |
135.000 |
| C1 |
C4 |
C3 |
90.000 |
|
C1 |
C4 |
F8 |
135.000 |
| C2 |
C1 |
C4 |
90.000 |
|
C2 |
C1 |
F5 |
135.000 |
| C2 |
C3 |
C4 |
90.000 |
|
C2 |
C3 |
F7 |
135.000 |
| C3 |
C2 |
F6 |
135.000 |
|
C3 |
C4 |
F8 |
135.000 |
| C4 |
C1 |
F5 |
135.000 |
|
C4 |
C3 |
F7 |
135.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.120 |
|
|
|
| 2 |
C |
0.566 |
|
|
|
| 3 |
C |
0.120 |
|
|
|
| 4 |
C |
0.566 |
|
|
|
| 5 |
F |
-0.370 |
|
|
|
| 6 |
F |
-0.316 |
|
|
|
| 7 |
F |
-0.370 |
|
|
|
| 8 |
F |
-0.316 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.866 |
0.000 |
0.000 |
| y |
0.000 |
-51.962 |
0.000 |
| z |
0.000 |
0.000 |
-40.905 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.568 |
0.000 |
0.000 |
| y |
0.000 |
-11.077 |
0.000 |
| z |
0.000 |
0.000 |
5.509 |
|
| Polar |
|---|
| 3z2-r2 | 11.018 |
|---|
| x2-y2 | 11.096 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.424 |
0.000 |
0.000 |
| y |
0.000 |
5.698 |
0.000 |
| z |
0.000 |
0.000 |
1.940 |
<r2> (average value of r
2) Å
2
| <r2> |
248.519 |
| (<r2>)1/2 |
15.764 |
Jump to
S1C1
Energy calculated at HF/3-21G*
| | hartrees |
|---|
| Energy at 0K | -546.023130 |
| Energy at 298.15K | -546.023629 |
| HF Energy | -546.023130 |
| Nuclear repulsion energy | 350.916253 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HF/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2081 |
1878 |
0.00 |
|
|
|
| 2 |
Ag |
1316 |
1188 |
0.00 |
|
|
|
| 3 |
Ag |
652 |
588 |
0.00 |
|
|
|
| 4 |
Ag |
350 |
316 |
0.00 |
|
|
|
| 5 |
Ag |
237 |
214 |
0.00 |
|
|
|
| 6 |
Au |
1442 |
1302 |
324.35 |
|
|
|
| 7 |
Au |
953 |
860 |
38.24 |
|
|
|
| 8 |
Au |
759 |
685 |
0.00 |
|
|
|
| 9 |
Au |
218 |
197 |
0.63 |
|
|
|
| 10 |
Au |
180 |
163 |
0.00 |
|
|
|
| 11 |
Bg |
1556 |
1404 |
0.00 |
|
|
|
| 12 |
Bg |
800 |
722 |
0.00 |
|
|
|
| 13 |
Bg |
602 |
543 |
0.00 |
|
|
|
| 14 |
Bg |
533 |
481 |
0.00 |
|
|
|
| 15 |
Bu |
2057 |
1857 |
40.54 |
|
|
|
| 16 |
Bu |
1058 |
955 |
162.73 |
|
|
|
| 17 |
Bu |
313 |
283 |
13.20 |
|
|
|
| 18 |
Bu |
260 |
234 |
3.23 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 7682.6 cm
-1
Scaled (by 0.9026) Zero Point Vibrational Energy (zpe) 6934.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HF/3-21G*
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.799 |
0.652 |
| C2 |
0.000 |
-0.799 |
0.652 |
| C3 |
0.000 |
-0.799 |
-0.652 |
| C4 |
0.000 |
0.799 |
-0.652 |
| F5 |
0.000 |
1.676 |
1.645 |
| F6 |
-0.000 |
-1.676 |
1.645 |
| F7 |
-0.000 |
-1.676 |
-1.645 |
| F8 |
0.000 |
1.676 |
-1.645 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
| C1 | | 1.5978 | 2.0620 | 1.3034 | 1.3250 | 2.6669 | 3.3763 | 2.4582 |
C2 | 1.5978 | | 1.3034 | 2.0620 | 2.6669 | 1.3250 | 2.4582 | 3.3763 | C3 | 2.0620 | 1.3034 | | 1.5978 | 3.3763 | 2.4582 | 1.3250 | 2.6669 | C4 | 1.3034 | 2.0620 | 1.5978 | | 2.4582 | 3.3763 | 2.6669 | 1.3250 | F5 | 1.3250 | 2.6669 | 3.3763 | 2.4582 | | 3.3526 | 4.6967 | 3.2892 | F6 | 2.6669 | 1.3250 | 2.4582 | 3.3763 | 3.3526 | | 3.2892 | 4.6967 | F7 | 3.3763 | 2.4582 | 1.3250 | 2.6669 | 4.6967 | 3.2892 | | 3.3526 | F8 | 2.4582 | 3.3763 | 2.6669 | 1.3250 | 3.2892 | 4.6967 | 3.3526 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
90.000 |
|
C1 |
C2 |
F6 |
131.465 |
| C1 |
C4 |
C3 |
90.000 |
|
C1 |
C4 |
F8 |
138.535 |
| C2 |
C1 |
C4 |
90.000 |
|
C2 |
C1 |
F5 |
131.465 |
| C2 |
C3 |
C4 |
90.000 |
|
C2 |
C3 |
F7 |
138.535 |
| C3 |
C2 |
F6 |
138.535 |
|
C3 |
C4 |
F8 |
131.465 |
| C4 |
C1 |
F5 |
138.535 |
|
C4 |
C3 |
F7 |
131.465 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.351 |
|
|
|
| 2 |
C |
0.351 |
|
|
|
| 3 |
C |
0.351 |
|
|
|
| 4 |
C |
0.351 |
|
|
|
| 5 |
F |
-0.351 |
|
|
|
| 6 |
F |
-0.351 |
|
|
|
| 7 |
F |
-0.351 |
|
|
|
| 8 |
F |
-0.351 |
|
|
|
Electric dipole moments
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.573 |
-0.002 |
0.000 |
| y |
-0.002 |
-46.255 |
0.000 |
| z |
0.000 |
0.000 |
-46.707 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.908 |
-0.002 |
0.000 |
| y |
-0.002 |
-2.615 |
0.000 |
| z |
0.000 |
0.000 |
-3.293 |
|
| Polar |
|---|
| 3z2-r2 | -6.586 |
|---|
| x2-y2 | 5.682 |
|---|
| xy | -0.002 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.948 |
0.000 |
0.000 |
| y |
0.000 |
4.636 |
0.000 |
| z |
0.000 |
0.000 |
6.644 |
<r2> (average value of r
2) Å
2
| <r2> |
251.842 |
| (<r2>)1/2 |
15.870 |