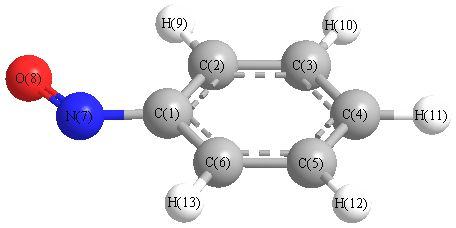
Vibrational Frequencies calculated at HF/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3417 |
3084 |
2.47 |
75.37 |
0.17 |
0.29 |
| 2 |
A' |
3397 |
3066 |
5.02 |
155.47 |
0.12 |
0.21 |
| 3 |
A' |
3385 |
3055 |
10.24 |
48.87 |
0.24 |
0.39 |
| 4 |
A' |
3376 |
3047 |
7.57 |
94.41 |
0.74 |
0.85 |
| 5 |
A' |
3362 |
3034 |
0.61 |
49.61 |
0.75 |
0.85 |
| 6 |
A' |
1770 |
1598 |
3.55 |
52.29 |
0.54 |
0.70 |
| 7 |
A' |
1762 |
1591 |
3.34 |
13.01 |
0.74 |
0.85 |
| 8 |
A' |
1662 |
1500 |
23.05 |
2.58 |
0.40 |
0.57 |
| 9 |
A' |
1638 |
1478 |
26.20 |
20.94 |
0.31 |
0.47 |
| 10 |
A' |
1588 |
1434 |
80.48 |
82.00 |
0.32 |
0.48 |
| 11 |
A' |
1488 |
1343 |
10.89 |
3.07 |
0.37 |
0.54 |
| 12 |
A' |
1370 |
1237 |
8.73 |
0.29 |
0.59 |
0.74 |
| 13 |
A' |
1318 |
1190 |
0.23 |
2.68 |
0.75 |
0.85 |
| 14 |
A' |
1266 |
1142 |
12.57 |
9.05 |
0.66 |
0.80 |
| 15 |
A' |
1232 |
1112 |
73.67 |
61.06 |
0.24 |
0.38 |
| 16 |
A' |
1188 |
1072 |
2.40 |
0.36 |
0.21 |
0.34 |
| 17 |
A' |
1132 |
1022 |
6.46 |
1.61 |
0.18 |
0.31 |
| 18 |
A' |
1103 |
995 |
0.16 |
35.74 |
0.12 |
0.21 |
| 19 |
A' |
870 |
785 |
21.39 |
8.40 |
0.59 |
0.74 |
| 20 |
A' |
728 |
657 |
19.90 |
3.56 |
0.27 |
0.43 |
| 21 |
A' |
705 |
636 |
0.03 |
6.00 |
0.75 |
0.86 |
| 22 |
A' |
492 |
444 |
0.07 |
2.81 |
0.31 |
0.47 |
| 23 |
A' |
283 |
255 |
3.22 |
2.65 |
0.51 |
0.67 |
| 24 |
A" |
1216 |
1098 |
1.88 |
1.48 |
0.75 |
0.86 |
| 25 |
A" |
1193 |
1077 |
0.03 |
0.12 |
0.75 |
0.86 |
| 26 |
A" |
1143 |
1032 |
10.29 |
2.02 |
0.75 |
0.86 |
| 27 |
A" |
1011 |
912 |
0.00 |
4.64 |
0.75 |
0.86 |
| 28 |
A" |
903 |
815 |
68.15 |
0.26 |
0.75 |
0.86 |
| 29 |
A" |
805 |
726 |
56.73 |
0.75 |
0.75 |
0.86 |
| 30 |
A" |
555 |
501 |
5.19 |
0.39 |
0.75 |
0.86 |
| 31 |
A" |
481 |
434 |
0.01 |
0.09 |
0.75 |
0.86 |
| 32 |
A" |
287 |
259 |
0.05 |
5.44 |
0.75 |
0.86 |
| 33 |
A" |
132 |
120 |
0.34 |
0.33 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 23127.8 cm
-1
Scaled (by 0.9026) Zero Point Vibrational Energy (zpe) 20875.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.139 |
|
|
|
| 2 |
C |
-0.188 |
|
|
|
| 3 |
C |
-0.248 |
|
|
|
| 4 |
C |
-0.216 |
|
|
|
| 5 |
C |
-0.249 |
|
|
|
| 6 |
C |
-0.180 |
|
|
|
| 7 |
N |
-0.046 |
|
|
|
| 8 |
O |
-0.345 |
|
|
|
| 9 |
H |
0.288 |
|
|
|
| 10 |
H |
0.256 |
|
|
|
| 11 |
H |
0.256 |
|
|
|
| 12 |
H |
0.255 |
|
|
|
| 13 |
H |
0.277 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.377 |
-3.505 |
0.000 |
3.766 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.126 |
3.179 |
0.000 |
| y |
3.179 |
-48.790 |
0.000 |
| z |
0.000 |
0.000 |
-49.155 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.846 |
3.179 |
0.000 |
| y |
3.179 |
-3.149 |
0.000 |
| z |
0.000 |
0.000 |
-3.697 |
|
| Polar |
|---|
| 3z2-r2 | -7.394 |
|---|
| x2-y2 | 6.664 |
|---|
| xy | 3.179 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.065 |
-1.493 |
0.000 |
| y |
-1.493 |
12.048 |
0.000 |
| z |
0.000 |
0.000 |
2.576 |
<r2> (average value of r
2) Å
2
| <r2> |
247.119 |
| (<r2>)1/2 |
15.720 |