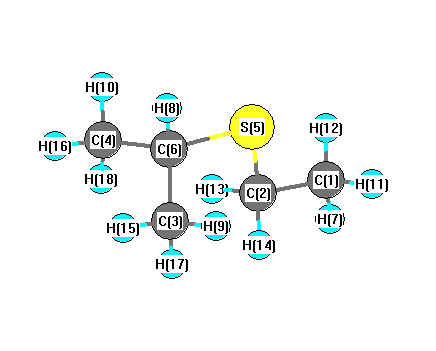
Vibrational Frequencies calculated at HF/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3287 |
2968 |
44.78 |
|
|
|
| 2 |
A |
3267 |
2950 |
49.42 |
|
|
|
| 3 |
A |
3261 |
2945 |
57.42 |
|
|
|
| 4 |
A |
3249 |
2934 |
4.97 |
|
|
|
| 5 |
A |
3238 |
2923 |
3.37 |
|
|
|
| 6 |
A |
3195 |
2885 |
38.29 |
|
|
|
| 7 |
A |
3190 |
2881 |
49.67 |
|
|
|
| 8 |
A |
1664 |
1502 |
9.23 |
|
|
|
| 9 |
A |
1660 |
1498 |
12.14 |
|
|
|
| 10 |
A |
1658 |
1497 |
8.37 |
|
|
|
| 11 |
A |
1648 |
1488 |
2.30 |
|
|
|
| 12 |
A |
1589 |
1435 |
5.47 |
|
|
|
| 13 |
A |
1584 |
1430 |
4.39 |
|
|
|
| 14 |
A |
1444 |
1303 |
32.65 |
|
|
|
| 15 |
A |
1425 |
1287 |
29.45 |
|
|
|
| 16 |
A |
1315 |
1188 |
11.79 |
|
|
|
| 17 |
A |
1215 |
1097 |
30.71 |
|
|
|
| 18 |
A |
1171 |
1057 |
0.49 |
|
|
|
| 19 |
A |
1074 |
970 |
5.67 |
|
|
|
| 20 |
A |
964 |
871 |
3.19 |
|
|
|
| 21 |
A |
705 |
636 |
7.72 |
|
|
|
| 22 |
A |
626 |
565 |
7.58 |
|
|
|
| 23 |
A |
496 |
448 |
1.96 |
|
|
|
| 24 |
A |
402 |
363 |
0.40 |
|
|
|
| 25 |
A |
308 |
278 |
1.49 |
|
|
|
| 26 |
A |
266 |
240 |
0.03 |
|
|
|
| 27 |
A |
162 |
146 |
0.28 |
|
|
|
| 28 |
A |
3316 |
2994 |
29.06 |
|
|
|
| 29 |
A |
3282 |
2963 |
29.13 |
|
|
|
| 30 |
A |
3276 |
2958 |
6.41 |
|
|
|
| 31 |
A |
3247 |
2932 |
1.09 |
|
|
|
| 32 |
A |
3184 |
2875 |
20.00 |
|
|
|
| 33 |
A |
1652 |
1491 |
4.65 |
|
|
|
| 34 |
A |
1650 |
1489 |
6.58 |
|
|
|
| 35 |
A |
1644 |
1484 |
0.66 |
|
|
|
| 36 |
A |
1572 |
1419 |
11.34 |
|
|
|
| 37 |
A |
1486 |
1342 |
0.42 |
|
|
|
| 38 |
A |
1406 |
1269 |
0.07 |
|
|
|
| 39 |
A |
1234 |
1114 |
2.37 |
|
|
|
| 40 |
A |
1157 |
1045 |
0.03 |
|
|
|
| 41 |
A |
1062 |
958 |
0.09 |
|
|
|
| 42 |
A |
1044 |
943 |
1.54 |
|
|
|
| 43 |
A |
878 |
793 |
4.43 |
|
|
|
| 44 |
A |
341 |
308 |
1.81 |
|
|
|
| 45 |
A |
251 |
227 |
0.05 |
|
|
|
| 46 |
A |
244 |
220 |
0.04 |
|
|
|
| 47 |
A |
69 |
63 |
2.10 |
|
|
|
| 48 |
A |
37 |
34 |
0.91 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 38047.3 cm
-1
Scaled (by 0.9029) Zero Point Vibrational Energy (zpe) 34352.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HF/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.454 |
-0.115 |
|
|
| 2 |
C |
-0.538 |
0.238 |
|
|
| 3 |
C |
-0.432 |
-0.253 |
|
|
| 4 |
C |
-0.432 |
-0.253 |
|
|
| 5 |
S |
0.206 |
-0.505 |
|
|
| 6 |
C |
-0.422 |
0.385 |
|
|
| 7 |
H |
0.162 |
0.006 |
|
|
| 8 |
H |
0.204 |
0.036 |
|
|
| 9 |
H |
0.177 |
0.059 |
|
|
| 10 |
H |
0.177 |
0.059 |
|
|
| 11 |
H |
0.174 |
0.058 |
|
|
| 12 |
H |
0.174 |
0.058 |
|
|
| 13 |
H |
0.189 |
-0.006 |
|
|
| 14 |
H |
0.189 |
-0.006 |
|
|
| 15 |
H |
0.159 |
0.044 |
|
|
| 16 |
H |
0.159 |
0.044 |
|
|
| 17 |
H |
0.155 |
0.075 |
|
|
| 18 |
H |
0.155 |
0.075 |
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.007 |
-2.196 |
0.000 |
2.196 |
| CHELPG |
0.003 |
-2.111 |
0.000 |
2.111 |
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-43.089 |
0.370 |
0.000 |
| y |
0.370 |
-50.998 |
0.000 |
| z |
0.000 |
0.000 |
-48.816 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.818 |
0.370 |
0.000 |
| y |
0.370 |
-5.046 |
0.000 |
| z |
0.000 |
0.000 |
-1.772 |
|
| Polar |
|---|
| 3z2-r2 | -3.544 |
|---|
| x2-y2 | 7.909 |
|---|
| xy | 0.370 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.626 |
0.986 |
0.000 |
| y |
0.986 |
9.624 |
0.000 |
| z |
0.000 |
0.000 |
8.514 |
<r2> (average value of r
2) Å
2
| <r2> |
274.509 |
| (<r2>)1/2 |
16.568 |