Vibrational Frequencies calculated at HF/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3484 |
3146 |
3.89 |
|
|
|
| 2 |
A |
3470 |
3133 |
0.46 |
|
|
|
| 3 |
A |
3288 |
2968 |
20.10 |
|
|
|
| 4 |
A |
3268 |
2950 |
17.83 |
|
|
|
| 5 |
A |
3202 |
2891 |
25.38 |
|
|
|
| 6 |
A |
1767 |
1596 |
21.57 |
|
|
|
| 7 |
A |
1674 |
1511 |
69.59 |
|
|
|
| 8 |
A |
1642 |
1483 |
10.61 |
|
|
|
| 9 |
A |
1635 |
1476 |
7.84 |
|
|
|
| 10 |
A |
1589 |
1435 |
5.61 |
|
|
|
| 11 |
A |
1450 |
1309 |
16.13 |
|
|
|
| 12 |
A |
1358 |
1226 |
3.62 |
|
|
|
| 13 |
A |
1270 |
1147 |
6.47 |
|
|
|
| 14 |
A |
1214 |
1096 |
10.08 |
|
|
|
| 15 |
A |
1141 |
1030 |
6.37 |
|
|
|
| 16 |
A |
1000 |
903 |
17.30 |
|
|
|
| 17 |
A |
974 |
879 |
53.58 |
|
|
|
| 18 |
A |
911 |
822 |
17.80 |
|
|
|
| 19 |
A |
889 |
803 |
38.96 |
|
|
|
| 20 |
A |
843 |
762 |
4.73 |
|
|
|
| 21 |
A |
728 |
657 |
0.93 |
|
|
|
| 22 |
A |
694 |
627 |
7.64 |
|
|
|
| 23 |
A |
585 |
528 |
7.07 |
|
|
|
| 24 |
A |
505 |
456 |
9.99 |
|
|
|
| 25 |
A |
356 |
322 |
2.71 |
|
|
|
| 26 |
A |
253 |
229 |
4.99 |
|
|
|
| 27 |
A |
142 |
128 |
0.35 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 19665.2 cm
-1
Scaled (by 0.9029) Zero Point Vibrational Energy (zpe) 17755.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
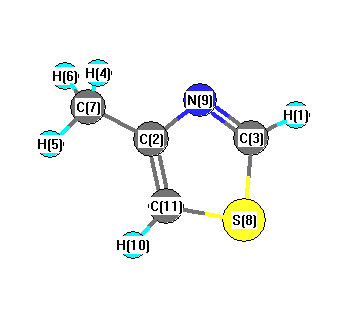
Charges from optimized geometry at HF/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.243 |
0.095 |
|
|
| 2 |
C |
0.269 |
0.607 |
|
|
| 3 |
C |
-0.243 |
0.343 |
|
|
| 4 |
H |
0.188 |
0.177 |
|
|
| 5 |
H |
0.163 |
0.121 |
|
|
| 6 |
H |
0.188 |
0.176 |
|
|
| 7 |
C |
-0.450 |
-0.581 |
|
|
| 8 |
S |
0.444 |
-0.128 |
|
|
| 9 |
N |
-0.443 |
-0.672 |
|
|
| 10 |
H |
0.238 |
0.234 |
|
|
| 11 |
C |
-0.599 |
-0.371 |
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.262 |
-1.123 |
-0.000 |
1.153 |
| CHELPG |
-0.172 |
-1.067 |
0.001 |
1.081 |
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.789 |
5.379 |
-0.001 |
| y |
5.379 |
-42.021 |
-0.000 |
| z |
-0.001 |
-0.000 |
-45.628 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.036 |
5.379 |
-0.001 |
| y |
5.379 |
1.187 |
-0.000 |
| z |
-0.001 |
-0.000 |
-4.223 |
|
| Polar |
|---|
| 3z2-r2 | -8.447 |
|---|
| x2-y2 | 1.232 |
|---|
| xy | 5.379 |
|---|
| xz | -0.001 |
|---|
| yz | -0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.929 |
-0.377 |
-0.000 |
| y |
-0.377 |
8.952 |
0.000 |
| z |
-0.000 |
0.000 |
4.440 |
<r2> (average value of r
2) Å
2
| <r2> |
183.003 |
| (<r2>)1/2 |
13.528 |