Jump to
S1C2
S2C1
S2C2
Energy calculated at CCSD/TZVP
| | hartrees |
|---|
| Energy at 0K | -131.105334 |
| Energy at 298.15K | -131.104728 |
| HF Energy | -130.720645 |
| Nuclear repulsion energy | 47.242294 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3371 |
3217 |
20.12 |
|
|
|
| 2 |
A' |
1853 |
1769 |
36.71 |
|
|
|
| 3 |
A' |
1156 |
1103 |
11.18 |
|
|
|
| 4 |
A' |
649 |
619 |
25.71 |
|
|
|
| 5 |
A' |
385 |
368 |
3.08 |
|
|
|
| 6 |
A" |
414 |
395 |
0.81 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3914.2 cm
-1
Scaled (by 0.9544) Zero Point Vibrational Energy (zpe) 3735.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CCSD/TZVP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.073 |
-1.236 |
0.000 |
| C2 |
0.000 |
0.095 |
0.000 |
| N3 |
-0.172 |
1.271 |
0.000 |
| H4 |
0.764 |
-2.054 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.3333 | 2.5190 | 1.0716 |
C2 | 1.3333 | | 1.1883 | 2.2816 | N3 | 2.5190 | 1.1883 | | 3.4548 | H4 | 1.0716 | 2.2816 | 3.4548 | |
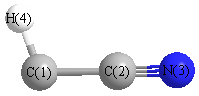 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
174.826 |
|
C2 |
C1 |
H4 |
142.922 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S2C1
S2C2
Energy calculated at CCSD/TZVP
| | hartrees |
|---|
| Energy at 0K | -131.102619 |
| Energy at 298.15K | |
| HF Energy | -130.718662 |
| Nuclear repulsion energy | 47.387234 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
3462 |
3304 |
65.28 |
|
|
|
| 2 |
Σ |
1669 |
1593 |
43.00 |
|
|
|
| 3 |
Σ |
1243 |
1187 |
6.04 |
|
|
|
| 4 |
Π |
404 |
386 |
0.15 |
|
|
|
| 4 |
Π |
404 |
386 |
0.15 |
|
|
|
| 5 |
Π |
552i |
527i |
55.27 |
|
|
|
| 5 |
Π |
552i |
527i |
55.27 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3039.1 cm
-1
Scaled (by 0.9544) Zero Point Vibrational Energy (zpe) 2900.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CCSD/TZVP
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.212 |
| C2 |
0.000 |
0.000 |
0.086 |
| N3 |
0.000 |
0.000 |
1.290 |
| H4 |
0.000 |
0.000 |
-2.276 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.2974 | 2.5019 | 1.0639 |
C2 | 1.2974 | | 1.2045 | 2.3613 | N3 | 2.5019 | 1.2045 | | 3.5658 | H4 | 1.0639 | 2.3613 | 3.5658 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
180.000 |
|
C2 |
C1 |
H4 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at CCSD/TZVP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.170 |
|
|
|
| 2 |
C |
0.007 |
|
|
|
| 3 |
N |
-0.048 |
|
|
|
| 4 |
H |
0.211 |
|
|
|
Electric dipole moments
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
36.259 |
| (<r2>)1/2 |
6.022 |
Jump to
S1C1
S1C2
S2C2
Energy calculated at CCSD/TZVP
| | hartrees |
|---|
| Energy at 0K | -131.078946 |
| Energy at 298.15K | -131.078347 |
| HF Energy | -130.657443 |
| Nuclear repulsion energy | 46.847652 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3076 |
2935 |
16.65 |
|
|
|
| 2 |
A' |
2157 |
2059 |
39.44 |
|
|
|
| 3 |
A' |
1065 |
1017 |
64.03 |
|
|
|
| 4 |
A' |
983 |
938 |
8.86 |
|
|
|
| 5 |
A' |
408 |
390 |
17.58 |
|
|
|
| 6 |
A" |
305 |
292 |
8.10 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3997.5 cm
-1
Scaled (by 0.9544) Zero Point Vibrational Energy (zpe) 3815.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at CCSD/TZVP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.061 |
-1.300 |
0.000 |
| C2 |
0.000 |
0.102 |
0.000 |
| N3 |
-0.213 |
1.253 |
0.000 |
| H4 |
1.124 |
-1.582 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.4031 | 2.5679 | 1.1002 |
C2 | 1.4031 | | 1.1710 | 2.0248 | N3 | 2.5679 | 1.1710 | | 3.1350 | H4 | 1.1002 | 2.0248 | 3.1350 | |
More geometry information
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
S2C1
Energy calculated at CCSD/TZVP
| | hartrees |
|---|
| Energy at 0K | -131.078946 |
| Energy at 298.15K | -131.078347 |
| HF Energy | -130.657443 |
| Nuclear repulsion energy | 46.847652 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at CCSD/TZVP
Geometric Data calculated at CCSD/TZVP
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability