Jump to
S1C2
Energy calculated at CCSD(T)/TZVP
| | hartrees |
|---|
| Energy at 0K | -412.658203 |
| Energy at 298.15K | |
| HF Energy | -412.503065 |
| Nuclear repulsion energy | 25.527030 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at CCSD(T)/TZVP
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
0.000 |
0.000 |
| Li2 |
0.000 |
0.000 |
2.083 |
| Li3 |
0.000 |
0.000 |
-2.083 |
Atom - Atom Distances (Å)
| |
S1 |
Li2 |
Li3 |
| S1 | | 2.0832 | 2.0832 |
Li2 | 2.0832 | | 4.1664 | Li3 | 2.0832 | 4.1664 | |
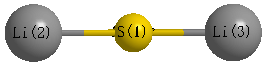 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li2 |
S1 |
Li3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at CCSD(T)/TZVP
| | hartrees |
|---|
| Energy at 0K | -412.659849 |
| Energy at 298.15K | -412.660150 |
| HF Energy | -412.501129 |
| Nuclear repulsion energy | 25.460669 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at CCSD(T)/TZVP
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.000 |
0.000 |
0.295 |
| Li2 |
0.000 |
1.804 |
-0.787 |
| Li3 |
0.000 |
-1.804 |
-0.787 |
Atom - Atom Distances (Å)
| |
S1 |
Li2 |
Li3 |
| S1 | | 2.1043 | 2.1043 |
Li2 | 2.1043 | | 3.6089 | Li3 | 2.1043 | 3.6089 | |
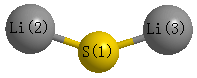 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li2 |
S1 |
Li3 |
118.073 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability