Vibrational Frequencies calculated at BLYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3036 |
3012 |
29.06 |
|
|
|
| 2 |
A' |
3033 |
3008 |
41.13 |
|
|
|
| 3 |
A' |
2976 |
2952 |
29.78 |
|
|
|
| 4 |
A' |
2969 |
2945 |
59.42 |
|
|
|
| 5 |
A' |
2965 |
2941 |
16.80 |
|
|
|
| 6 |
A' |
2964 |
2940 |
29.94 |
|
|
|
| 7 |
A' |
2954 |
2930 |
14.45 |
|
|
|
| 8 |
A' |
1502 |
1490 |
2.07 |
|
|
|
| 9 |
A' |
1499 |
1487 |
1.26 |
|
|
|
| 10 |
A' |
1486 |
1474 |
0.46 |
|
|
|
| 11 |
A' |
1481 |
1469 |
2.07 |
|
|
|
| 12 |
A' |
1473 |
1461 |
9.87 |
|
|
|
| 13 |
A' |
1402 |
1391 |
0.72 |
|
|
|
| 14 |
A' |
1399 |
1387 |
0.87 |
|
|
|
| 15 |
A' |
1351 |
1340 |
0.67 |
|
|
|
| 16 |
A' |
1283 |
1273 |
22.66 |
|
|
|
| 17 |
A' |
1234 |
1224 |
53.48 |
|
|
|
| 18 |
A' |
1093 |
1084 |
2.58 |
|
|
|
| 19 |
A' |
1042 |
1034 |
5.81 |
|
|
|
| 20 |
A' |
1012 |
1004 |
0.14 |
|
|
|
| 21 |
A' |
964 |
956 |
3.92 |
|
|
|
| 22 |
A' |
881 |
874 |
1.95 |
|
|
|
| 23 |
A' |
718 |
712 |
2.76 |
|
|
|
| 24 |
A' |
643 |
638 |
1.85 |
|
|
|
| 25 |
A' |
369 |
366 |
0.38 |
|
|
|
| 26 |
A' |
285 |
282 |
0.70 |
|
|
|
| 27 |
A' |
264 |
262 |
0.85 |
|
|
|
| 28 |
A' |
101 |
100 |
0.33 |
|
|
|
| 29 |
A" |
3049 |
3025 |
33.35 |
|
|
|
| 30 |
A" |
3031 |
3006 |
71.50 |
|
|
|
| 31 |
A" |
3007 |
2982 |
2.47 |
|
|
|
| 32 |
A" |
3003 |
2978 |
25.30 |
|
|
|
| 33 |
A" |
2986 |
2962 |
1.01 |
|
|
|
| 34 |
A" |
1494 |
1482 |
5.50 |
|
|
|
| 35 |
A" |
1485 |
1473 |
6.01 |
|
|
|
| 36 |
A" |
1302 |
1291 |
0.14 |
|
|
|
| 37 |
A" |
1245 |
1235 |
0.03 |
|
|
|
| 38 |
A" |
1222 |
1212 |
0.01 |
|
|
|
| 39 |
A" |
1048 |
1039 |
1.40 |
|
|
|
| 40 |
A" |
1020 |
1012 |
0.08 |
|
|
|
| 41 |
A" |
858 |
851 |
0.11 |
|
|
|
| 42 |
A" |
781 |
774 |
5.06 |
|
|
|
| 43 |
A" |
740 |
734 |
3.21 |
|
|
|
| 44 |
A" |
243 |
241 |
0.03 |
|
|
|
| 45 |
A" |
239 |
237 |
0.06 |
|
|
|
| 46 |
A" |
113 |
112 |
0.87 |
|
|
|
| 47 |
A" |
63 |
62 |
0.40 |
|
|
|
| 48 |
A" |
38 |
38 |
0.26 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 34671.8 cm
-1
Scaled (by 0.9919) Zero Point Vibrational Energy (zpe) 34390.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
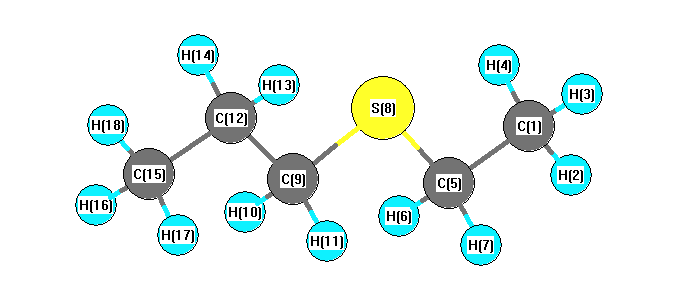
Charges from optimized geometry at BLYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.412 |
|
|
|
| 2 |
H |
0.136 |
|
|
|
| 3 |
H |
0.150 |
|
|
|
| 4 |
H |
0.150 |
|
|
|
| 5 |
C |
-0.351 |
|
|
|
| 6 |
H |
0.149 |
|
|
|
| 7 |
H |
0.149 |
|
|
|
| 8 |
S |
0.057 |
|
|
|
| 9 |
C |
-0.357 |
|
|
|
| 10 |
H |
0.145 |
|
|
|
| 11 |
H |
0.145 |
|
|
|
| 12 |
C |
-0.223 |
|
|
|
| 13 |
H |
0.136 |
|
|
|
| 14 |
H |
0.136 |
|
|
|
| 15 |
C |
-0.406 |
|
|
|
| 16 |
H |
0.131 |
|
|
|
| 17 |
H |
0.131 |
|
|
|
| 18 |
H |
0.135 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.958 |
-1.308 |
0.000 |
1.621 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-43.547 |
2.500 |
0.000 |
| y |
2.500 |
-47.160 |
0.000 |
| z |
0.000 |
0.000 |
-47.923 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.994 |
2.500 |
0.000 |
| y |
2.500 |
-1.426 |
0.000 |
| z |
0.000 |
0.000 |
-2.569 |
|
| Polar |
|---|
| 3z2-r2 | -5.138 |
|---|
| x2-y2 | 3.613 |
|---|
| xy | 2.500 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.519 |
2.594 |
0.000 |
| y |
2.594 |
12.064 |
0.000 |
| z |
0.000 |
0.000 |
8.654 |
<r2> (average value of r
2) Å
2
| <r2> |
364.061 |
| (<r2>)1/2 |
19.080 |