Jump to
S1C2
Vibrational Frequencies calculated at BLYP/6-31G**
Geometric Data calculated at BLYP/6-31G**
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at BLYP/6-31G**
| | hartrees |
|---|
| Energy at 0K | -271.657910 |
| Energy at 298.15K | -271.668144 |
| Nuclear repulsion energy | 233.193450 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at BLYP/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3084 |
3060 |
13.40 |
|
|
|
| 2 |
A |
3033 |
3010 |
42.25 |
|
|
|
| 3 |
A |
3030 |
3006 |
71.87 |
|
|
|
| 4 |
A |
3023 |
3000 |
8.96 |
|
|
|
| 5 |
A |
3005 |
2982 |
0.03 |
|
|
|
| 6 |
A |
2980 |
2957 |
22.89 |
|
|
|
| 7 |
A |
2968 |
2945 |
6.89 |
|
|
|
| 8 |
A |
2961 |
2938 |
28.88 |
|
|
|
| 9 |
A |
2954 |
2932 |
10.64 |
|
|
|
| 10 |
A |
2931 |
2909 |
15.53 |
|
|
|
| 11 |
A |
1725 |
1712 |
114.05 |
|
|
|
| 12 |
A |
1486 |
1474 |
2.87 |
|
|
|
| 13 |
A |
1479 |
1467 |
4.31 |
|
|
|
| 14 |
A |
1465 |
1454 |
2.37 |
|
|
|
| 15 |
A |
1456 |
1444 |
8.62 |
|
|
|
| 16 |
A |
1443 |
1432 |
13.68 |
|
|
|
| 17 |
A |
1427 |
1416 |
2.97 |
|
|
|
| 18 |
A |
1391 |
1380 |
0.73 |
|
|
|
| 19 |
A |
1357 |
1347 |
31.52 |
|
|
|
| 20 |
A |
1352 |
1342 |
18.07 |
|
|
|
| 21 |
A |
1293 |
1283 |
0.13 |
|
|
|
| 22 |
A |
1287 |
1278 |
12.40 |
|
|
|
| 23 |
A |
1222 |
1213 |
0.00 |
|
|
|
| 24 |
A |
1146 |
1137 |
81.61 |
|
|
|
| 25 |
A |
1110 |
1101 |
0.95 |
|
|
|
| 26 |
A |
1097 |
1088 |
0.06 |
|
|
|
| 27 |
A |
1016 |
1008 |
0.18 |
|
|
|
| 28 |
A |
953 |
945 |
1.04 |
|
|
|
| 29 |
A |
932 |
925 |
6.83 |
|
|
|
| 30 |
A |
873 |
867 |
13.42 |
|
|
|
| 31 |
A |
816 |
810 |
0.64 |
|
|
|
| 32 |
A |
789 |
783 |
1.94 |
|
|
|
| 33 |
A |
709 |
704 |
3.56 |
|
|
|
| 34 |
A |
571 |
566 |
9.43 |
|
|
|
| 35 |
A |
461 |
457 |
0.00 |
|
|
|
| 36 |
A |
383 |
380 |
1.07 |
|
|
|
| 37 |
A |
330 |
327 |
2.53 |
|
|
|
| 38 |
A |
243 |
241 |
0.04 |
|
|
|
| 39 |
A |
170 |
169 |
4.45 |
|
|
|
| 40 |
A |
107 |
106 |
0.11 |
|
|
|
| 41 |
A |
89 |
88 |
0.42 |
|
|
|
| 42 |
A |
22 |
22 |
0.51 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30082.9 cm
-1
Scaled (by 0.9923) Zero Point Vibrational Energy (zpe) 29851.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at BLYP/6-31G**
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
2.785 |
-0.291 |
-0.006 |
| C2 |
1.421 |
0.428 |
0.030 |
| C3 |
0.227 |
-0.544 |
-0.019 |
| C4 |
-1.147 |
0.145 |
-0.007 |
| C5 |
-2.373 |
-0.775 |
0.013 |
| O6 |
-1.263 |
1.369 |
-0.014 |
| H7 |
3.616 |
0.431 |
0.030 |
| H8 |
2.903 |
-0.978 |
0.849 |
| H9 |
2.903 |
-0.887 |
-0.927 |
| H10 |
1.343 |
1.045 |
0.941 |
| H11 |
1.341 |
1.134 |
-0.813 |
| H12 |
0.276 |
-1.181 |
-0.924 |
| H13 |
0.262 |
-1.258 |
0.827 |
| H14 |
-3.290 |
-0.173 |
-0.035 |
| H15 |
-2.348 |
-1.482 |
-0.834 |
| H16 |
-2.382 |
-1.383 |
0.934 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
| C1 | | 1.5419 | 2.5697 | 3.9562 | 5.1807 | 4.3747 | 1.1017 | 1.1032 | 1.1032 | 2.1822 | 2.1831 | 2.8154 | 2.8274 | 6.0761 | 5.3336 | 5.3633 |
C2 | 1.5419 | | 1.5402 | 2.5846 | 3.9810 | 2.8446 | 2.1941 | 2.2008 | 2.2002 | 1.1029 | 1.1025 | 2.1934 | 2.1961 | 4.7504 | 4.3132 | 4.3080 | C3 | 2.5697 | 1.5402 | | 1.5377 | 2.6113 | 2.4246 | 3.5259 | 2.8464 | 2.8462 | 2.1657 | 2.1644 | 1.1083 | 1.1077 | 3.5371 | 2.8597 | 2.9013 | C4 | 3.9562 | 2.5846 | 1.5377 | | 1.5330 | 1.2293 | 4.7716 | 4.2899 | 4.2795 | 2.8129 | 2.7961 | 2.1505 | 2.1567 | 2.1666 | 2.1846 | 2.1781 | C5 | 5.1807 | 3.9810 | 2.6113 | 1.5330 | | 2.4148 | 6.1092 | 5.3465 | 5.3603 | 4.2412 | 4.2569 | 2.8393 | 2.8003 | 1.0977 | 1.1032 | 1.1037 | O6 | 4.3747 | 2.8446 | 2.4246 | 1.2293 | 2.4148 | | 4.9677 | 4.8589 | 4.8244 | 2.7940 | 2.7336 | 3.1142 | 3.1516 | 2.5476 | 3.1590 | 3.1183 | H7 | 1.1017 | 2.1941 | 3.5259 | 4.7716 | 6.1092 | 4.9677 | | 1.7786 | 1.7786 | 2.5239 | 2.5255 | 3.8290 | 3.8387 | 6.9325 | 6.3222 | 6.3303 | H8 | 1.1032 | 2.2008 | 2.8464 | 4.2899 | 5.3465 | 4.8589 | 1.7786 | | 1.7784 | 2.5567 | 3.1085 | 3.1759 | 2.6564 | 6.3079 | 5.5373 | 5.3011 | H9 | 1.1032 | 2.2002 | 2.8462 | 4.2795 | 5.3603 | 4.8244 | 1.7786 | 1.7784 | | 3.1076 | 2.5567 | 2.6429 | 3.1921 | 6.2975 | 5.2850 | 5.6241 | H10 | 2.1822 | 1.1029 | 2.1657 | 2.8129 | 4.2412 | 2.7940 | 2.5239 | 2.5567 | 3.1076 | | 1.7561 | 3.0939 | 2.5470 | 4.8893 | 4.8127 | 4.4464 | H11 | 2.1831 | 1.1025 | 2.1644 | 2.7961 | 4.2569 | 2.7336 | 2.5255 | 3.1085 | 2.5567 | 1.7561 | | 2.5503 | 3.0944 | 4.8746 | 4.5223 | 4.8210 | H12 | 2.8154 | 2.1934 | 1.1083 | 2.1505 | 2.8393 | 3.1142 | 3.8290 | 3.1759 | 2.6429 | 3.0939 | 2.5503 | | 1.7530 | 3.8111 | 2.6428 | 3.2487 | H13 | 2.8274 | 2.1961 | 1.1077 | 2.1567 | 2.8003 | 3.1516 | 3.8387 | 2.6564 | 3.1921 | 2.5470 | 3.0944 | 1.7530 | | 3.8128 | 3.1018 | 2.6485 | H14 | 6.0761 | 4.7504 | 3.5371 | 2.1666 | 1.0977 | 2.5476 | 6.9325 | 6.3079 | 6.2975 | 4.8893 | 4.8746 | 3.8111 | 3.8128 | | 1.7998 | 1.7965 | H15 | 5.3336 | 4.3132 | 2.8597 | 2.1846 | 1.1032 | 3.1590 | 6.3222 | 5.5373 | 5.2850 | 4.8127 | 4.5223 | 2.6428 | 3.1018 | 1.7998 | | 1.7705 | H16 | 5.3633 | 4.3080 | 2.9013 | 2.1781 | 1.1037 | 3.1183 | 6.3303 | 5.3011 | 5.6241 | 4.4464 | 4.8210 | 3.2487 | 2.6485 | 1.7965 | 1.7705 | |
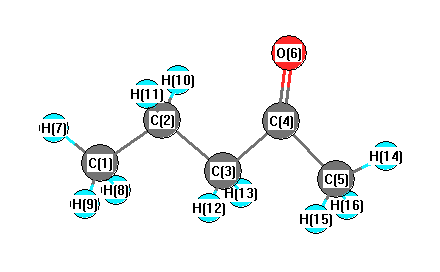 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
112.974 |
|
C1 |
C2 |
H10 |
110.092 |
| C1 |
C2 |
H11 |
110.186 |
|
C2 |
C1 |
H7 |
111.099 |
| C2 |
C1 |
H8 |
111.538 |
|
C2 |
C1 |
H9 |
111.495 |
| C2 |
C3 |
C4 |
114.224 |
|
C2 |
C3 |
H12 |
110.769 |
| C2 |
C3 |
H13 |
111.011 |
|
C3 |
C2 |
H10 |
108.915 |
| C3 |
C2 |
H11 |
108.845 |
|
C3 |
C4 |
C5 |
116.507 |
| C3 |
C4 |
O6 |
121.987 |
|
C4 |
C3 |
H12 |
107.622 |
| C4 |
C3 |
H13 |
108.129 |
|
C4 |
C5 |
H14 |
109.784 |
| C4 |
C5 |
H15 |
110.872 |
|
C4 |
C5 |
H16 |
110.333 |
| C5 |
C4 |
O6 |
121.506 |
|
H7 |
C1 |
H8 |
107.539 |
| H7 |
C1 |
H9 |
107.543 |
|
H8 |
C1 |
H9 |
107.417 |
| H10 |
C2 |
H11 |
105.549 |
|
H12 |
C3 |
H13 |
104.575 |
| H14 |
C5 |
H15 |
109.720 |
|
H14 |
C5 |
H16 |
109.388 |
| H15 |
C5 |
H16 |
106.688 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.290 |
|
|
|
| 2 |
H |
0.089 |
|
|
|
| 3 |
H |
0.089 |
|
|
|
| 4 |
H |
0.092 |
|
|
|
| 5 |
C |
-0.132 |
|
|
|
| 6 |
H |
0.096 |
|
|
|
| 7 |
H |
0.095 |
|
|
|
| 8 |
C |
-0.216 |
|
|
|
| 9 |
H |
0.098 |
|
|
|
| 10 |
C |
-0.350 |
|
|
|
| 11 |
H |
0.121 |
|
|
|
| 12 |
H |
0.113 |
|
|
|
| 13 |
C |
0.398 |
|
|
|
| 14 |
O |
-0.413 |
|
|
|
| 15 |
H |
0.098 |
|
|
|
| 16 |
H |
0.113 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.314 |
-2.500 |
0.001 |
2.520 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.513 |
-3.768 |
0.002 |
| y |
-3.768 |
-41.112 |
0.000 |
| z |
0.002 |
0.000 |
-37.017 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.552 |
-3.768 |
0.002 |
| y |
-3.768 |
-3.847 |
0.000 |
| z |
0.002 |
0.000 |
2.296 |
|
| Polar |
|---|
| 3z2-r2 | 4.591 |
|---|
| x2-y2 | 3.599 |
|---|
| xy | -3.768 |
|---|
| xz | 0.002 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
232.945 |
| (<r2>)1/2 |
15.263 |