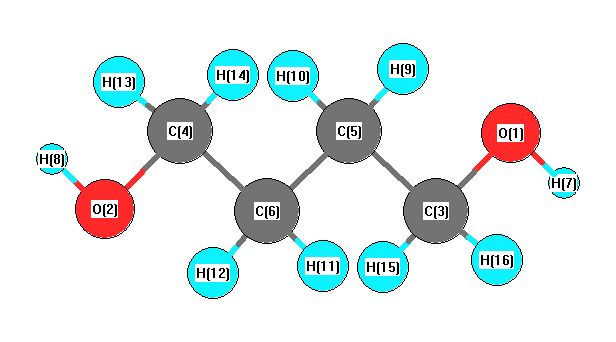
Vibrational Frequencies calculated at BLYP/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3679 |
3668 |
0.00 |
|
|
|
| 2 |
Ag |
2945 |
2936 |
0.00 |
|
|
|
| 3 |
Ag |
2890 |
2881 |
0.00 |
|
|
|
| 4 |
Ag |
1487 |
1483 |
0.00 |
|
|
|
| 5 |
Ag |
1460 |
1455 |
0.00 |
|
|
|
| 6 |
Ag |
1405 |
1400 |
0.00 |
|
|
|
| 7 |
Ag |
1341 |
1337 |
0.00 |
|
|
|
| 8 |
Ag |
1229 |
1225 |
0.00 |
|
|
|
| 9 |
Ag |
1018 |
1015 |
0.00 |
|
|
|
| 10 |
Ag |
1017 |
1014 |
0.00 |
|
|
|
| 11 |
Ag |
969 |
966 |
0.00 |
|
|
|
| 12 |
Ag |
348 |
347 |
0.00 |
|
|
|
| 13 |
Ag |
325 |
324 |
0.00 |
|
|
|
| 14 |
Au |
2994 |
2985 |
89.95 |
|
|
|
| 15 |
Au |
2909 |
2900 |
86.42 |
|
|
|
| 16 |
Au |
1287 |
1283 |
1.68 |
|
|
|
| 17 |
Au |
1179 |
1175 |
1.46 |
|
|
|
| 18 |
Au |
930 |
927 |
1.49 |
|
|
|
| 19 |
Au |
743 |
741 |
0.91 |
|
|
|
| 20 |
Au |
251 |
251 |
202.79 |
|
|
|
| 21 |
Au |
92 |
92 |
14.48 |
|
|
|
| 22 |
Au |
73 |
73 |
2.18 |
|
|
|
| 23 |
Bg |
2969 |
2960 |
0.00 |
|
|
|
| 24 |
Bg |
2911 |
2902 |
0.00 |
|
|
|
| 25 |
Bg |
1289 |
1285 |
0.00 |
|
|
|
| 26 |
Bg |
1246 |
1242 |
0.00 |
|
|
|
| 27 |
Bg |
1140 |
1137 |
0.00 |
|
|
|
| 28 |
Bg |
800 |
798 |
0.00 |
|
|
|
| 29 |
Bg |
249 |
248 |
0.00 |
|
|
|
| 30 |
Bg |
143 |
142 |
0.00 |
|
|
|
| 31 |
Bu |
3679 |
3668 |
27.77 |
|
|
|
| 32 |
Bu |
2955 |
2946 |
74.46 |
|
|
|
| 33 |
Bu |
2889 |
2881 |
111.34 |
|
|
|
| 34 |
Bu |
1494 |
1489 |
3.10 |
|
|
|
| 35 |
Bu |
1469 |
1465 |
2.93 |
|
|
|
| 36 |
Bu |
1408 |
1404 |
11.20 |
|
|
|
| 37 |
Bu |
1280 |
1276 |
49.35 |
|
|
|
| 38 |
Bu |
1179 |
1176 |
51.47 |
|
|
|
| 39 |
Bu |
994 |
991 |
200.71 |
|
|
|
| 40 |
Bu |
943 |
940 |
5.19 |
|
|
|
| 41 |
Bu |
504 |
503 |
38.74 |
|
|
|
| 42 |
Bu |
133 |
132 |
5.19 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30121.1 cm
-1
Scaled (by 0.997) Zero Point Vibrational Energy (zpe) 30030.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at BLYP/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.327 |
|
|
|
| 2 |
O |
-0.327 |
|
|
|
| 3 |
C |
0.027 |
|
|
|
| 4 |
C |
0.027 |
|
|
|
| 5 |
C |
-0.134 |
|
|
|
| 6 |
C |
-0.134 |
|
|
|
| 7 |
H |
0.187 |
|
|
|
| 8 |
H |
0.187 |
|
|
|
| 9 |
H |
0.078 |
|
|
|
| 10 |
H |
0.078 |
|
|
|
| 11 |
H |
0.078 |
|
|
|
| 12 |
H |
0.078 |
|
|
|
| 13 |
H |
0.046 |
|
|
|
| 14 |
H |
0.046 |
|
|
|
| 15 |
H |
0.046 |
|
|
|
| 16 |
H |
0.046 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.242 |
7.525 |
0.000 |
| y |
7.525 |
-44.103 |
0.000 |
| z |
0.000 |
0.000 |
-39.031 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
12.325 |
7.525 |
0.000 |
| y |
7.525 |
-9.966 |
0.000 |
| z |
0.000 |
0.000 |
-2.359 |
|
| Polar |
|---|
| 3z2-r2 | -4.718 |
|---|
| x2-y2 | 14.861 |
|---|
| xy | 7.525 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
290.616 |
| (<r2>)1/2 |
17.047 |