Vibrational Frequencies calculated at B3PW91/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3268 |
3152 |
1.44 |
|
|
|
| 2 |
A |
3235 |
3121 |
0.21 |
|
|
|
| 3 |
A |
3148 |
3037 |
11.83 |
|
|
|
| 4 |
A |
3117 |
3007 |
10.13 |
|
|
|
| 5 |
A |
3047 |
2939 |
21.78 |
|
|
|
| 6 |
A |
1579 |
1523 |
38.92 |
|
|
|
| 7 |
A |
1489 |
1436 |
39.40 |
|
|
|
| 8 |
A |
1458 |
1406 |
10.05 |
|
|
|
| 9 |
A |
1448 |
1397 |
7.10 |
|
|
|
| 10 |
A |
1391 |
1342 |
5.62 |
|
|
|
| 11 |
A |
1355 |
1307 |
9.98 |
|
|
|
| 12 |
A |
1244 |
1200 |
1.13 |
|
|
|
| 13 |
A |
1154 |
1113 |
3.65 |
|
|
|
| 14 |
A |
1044 |
1007 |
2.24 |
|
|
|
| 15 |
A |
1005 |
969 |
8.46 |
|
|
|
| 16 |
A |
952 |
919 |
12.54 |
|
|
|
| 17 |
A |
891 |
860 |
29.14 |
|
|
|
| 18 |
A |
837 |
807 |
11.18 |
|
|
|
| 19 |
A |
816 |
787 |
38.18 |
|
|
|
| 20 |
A |
730 |
704 |
11.92 |
|
|
|
| 21 |
A |
678 |
654 |
1.18 |
|
|
|
| 22 |
A |
653 |
630 |
0.09 |
|
|
|
| 23 |
A |
559 |
539 |
1.24 |
|
|
|
| 24 |
A |
490 |
472 |
3.03 |
|
|
|
| 25 |
A |
335 |
324 |
2.72 |
|
|
|
| 26 |
A |
235 |
227 |
4.38 |
|
|
|
| 27 |
A |
133 |
128 |
0.51 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 18145.1 cm
-1
Scaled (by 0.9646) Zero Point Vibrational Energy (zpe) 17502.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
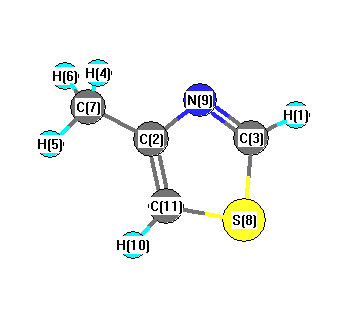
Charges from optimized geometry at B3PW91/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
-0.319 |
|
|
|
| 2 |
C |
0.123 |
|
|
|
| 3 |
C |
0.264 |
|
|
|
| 4 |
H |
-0.037 |
|
|
|
| 5 |
H |
-0.164 |
|
|
|
| 6 |
H |
-0.037 |
|
|
|
| 7 |
C |
0.404 |
|
|
|
| 8 |
S |
0.283 |
|
|
|
| 9 |
N |
-0.381 |
|
|
|
| 10 |
H |
-0.561 |
|
|
|
| 11 |
C |
0.426 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.119 |
-1.274 |
0.000 |
1.280 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-39.277 |
3.996 |
0.000 |
| y |
3.996 |
-41.386 |
0.000 |
| z |
0.000 |
0.000 |
-44.602 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.717 |
3.996 |
0.000 |
| y |
3.996 |
0.554 |
0.000 |
| z |
0.000 |
0.000 |
-4.271 |
|
| Polar |
|---|
| 3z2-r2 | -8.541 |
|---|
| x2-y2 | 2.109 |
|---|
| xy | 3.996 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.701 |
-0.278 |
0.000 |
| y |
-0.278 |
11.197 |
0.000 |
| z |
0.000 |
0.000 |
7.350 |
<r2> (average value of r
2) Å
2
| <r2> |
179.927 |
| (<r2>)1/2 |
13.414 |