Vibrational Frequencies calculated at B3PW91/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3198 |
3074 |
3.61 |
|
|
|
| 2 |
A |
3161 |
3039 |
8.65 |
|
|
|
| 3 |
A |
3138 |
3017 |
19.05 |
|
|
|
| 4 |
A |
3130 |
3009 |
20.74 |
|
|
|
| 5 |
A |
3099 |
2980 |
0.80 |
|
|
|
| 6 |
A |
3049 |
2931 |
25.40 |
|
|
|
| 7 |
A |
3031 |
2914 |
11.71 |
|
|
|
| 8 |
A |
2342 |
2251 |
17.88 |
|
|
|
| 9 |
A |
1697 |
1632 |
18.20 |
|
|
|
| 10 |
A |
1499 |
1441 |
5.88 |
|
|
|
| 11 |
A |
1492 |
1435 |
5.99 |
|
|
|
| 12 |
A |
1471 |
1414 |
5.08 |
|
|
|
| 13 |
A |
1418 |
1363 |
0.57 |
|
|
|
| 14 |
A |
1399 |
1345 |
0.70 |
|
|
|
| 15 |
A |
1331 |
1279 |
2.68 |
|
|
|
| 16 |
A |
1287 |
1238 |
0.01 |
|
|
|
| 17 |
A |
1250 |
1202 |
0.58 |
|
|
|
| 18 |
A |
1137 |
1093 |
0.60 |
|
|
|
| 19 |
A |
1087 |
1045 |
2.70 |
|
|
|
| 20 |
A |
1038 |
998 |
4.38 |
|
|
|
| 21 |
A |
1002 |
963 |
1.00 |
|
|
|
| 22 |
A |
967 |
930 |
4.50 |
|
|
|
| 23 |
A |
869 |
836 |
4.12 |
|
|
|
| 24 |
A |
804 |
773 |
6.76 |
|
|
|
| 25 |
A |
771 |
741 |
29.31 |
|
|
|
| 26 |
A |
674 |
648 |
2.29 |
|
|
|
| 27 |
A |
579 |
557 |
0.83 |
|
|
|
| 28 |
A |
402 |
386 |
0.36 |
|
|
|
| 29 |
A |
368 |
354 |
0.10 |
|
|
|
| 30 |
A |
244 |
235 |
0.80 |
|
|
|
| 31 |
A |
215 |
207 |
2.96 |
|
|
|
| 32 |
A |
144 |
138 |
4.83 |
|
|
|
| 33 |
A |
62 |
59 |
1.04 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23677.1 cm
-1
Scaled (by 0.9614) Zero Point Vibrational Energy (zpe) 22763.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
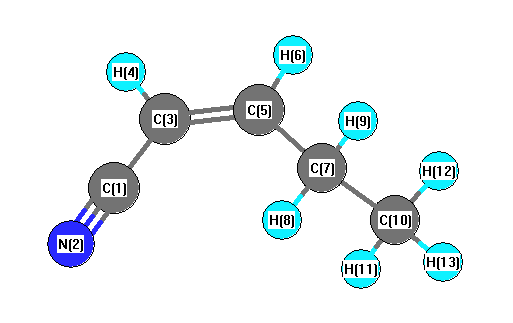
Charges from optimized geometry at B3PW91/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.300 |
|
|
|
| 2 |
N |
-0.362 |
|
|
|
| 3 |
C |
-0.233 |
|
|
|
| 4 |
H |
0.164 |
|
|
|
| 5 |
C |
-0.025 |
|
|
|
| 6 |
H |
0.142 |
|
|
|
| 7 |
C |
-0.309 |
|
|
|
| 8 |
H |
0.163 |
|
|
|
| 9 |
H |
0.151 |
|
|
|
| 10 |
C |
-0.444 |
|
|
|
| 11 |
H |
0.157 |
|
|
|
| 12 |
H |
0.144 |
|
|
|
| 13 |
H |
0.152 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
3.387 |
2.474 |
0.058 |
4.195 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-41.953 |
-5.543 |
0.972 |
| y |
-5.543 |
-35.298 |
-0.664 |
| z |
0.972 |
-0.664 |
-36.219 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.194 |
-5.543 |
0.972 |
| y |
-5.543 |
3.788 |
-0.664 |
| z |
0.972 |
-0.664 |
2.406 |
|
| Polar |
|---|
| 3z2-r2 | 4.813 |
|---|
| x2-y2 | -6.655 |
|---|
| xy | -5.543 |
|---|
| xz | 0.972 |
|---|
| yz | -0.664 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.909 |
0.982 |
0.533 |
| y |
0.982 |
8.351 |
-0.271 |
| z |
0.533 |
-0.271 |
5.853 |
<r2> (average value of r
2) Å
2
| <r2> |
200.836 |
| (<r2>)1/2 |
14.172 |