Jump to
S1C2
Energy calculated at B3PW91/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -578.725517 |
| Energy at 298.15K | -578.733129 |
| Nuclear repulsion energy | 158.599370 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3112 |
2993 |
25.79 |
|
|
|
| 2 |
A' |
3079 |
2961 |
25.32 |
|
|
|
| 3 |
A' |
3051 |
2933 |
9.57 |
|
|
|
| 4 |
A' |
3035 |
2918 |
21.08 |
|
|
|
| 5 |
A' |
1502 |
1445 |
7.92 |
|
|
|
| 6 |
A' |
1487 |
1430 |
0.72 |
|
|
|
| 7 |
A' |
1481 |
1424 |
1.75 |
|
|
|
| 8 |
A' |
1405 |
1351 |
2.98 |
|
|
|
| 9 |
A' |
1366 |
1313 |
5.84 |
|
|
|
| 10 |
A' |
1277 |
1228 |
20.24 |
|
|
|
| 11 |
A' |
1120 |
1077 |
0.59 |
|
|
|
| 12 |
A' |
1046 |
1006 |
1.60 |
|
|
|
| 13 |
A' |
910 |
875 |
14.07 |
|
|
|
| 14 |
A' |
745 |
716 |
36.26 |
|
|
|
| 15 |
A' |
360 |
347 |
2.79 |
|
|
|
| 16 |
A' |
230 |
221 |
1.88 |
|
|
|
| 17 |
A" |
3138 |
3017 |
19.58 |
|
|
|
| 18 |
A" |
3106 |
2987 |
28.46 |
|
|
|
| 19 |
A" |
3082 |
2963 |
0.00 |
|
|
|
| 20 |
A" |
1491 |
1434 |
8.17 |
|
|
|
| 21 |
A" |
1317 |
1266 |
0.00 |
|
|
|
| 22 |
A" |
1242 |
1194 |
0.22 |
|
|
|
| 23 |
A" |
1090 |
1049 |
1.00 |
|
|
|
| 24 |
A" |
868 |
835 |
0.14 |
|
|
|
| 25 |
A" |
749 |
720 |
3.88 |
|
|
|
| 26 |
A" |
219 |
211 |
0.02 |
|
|
|
| 27 |
A" |
117 |
113 |
1.45 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20810.9 cm
-1
Scaled (by 0.9616) Zero Point Vibrational Energy (zpe) 20011.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.583 |
0.000 |
| C2 |
0.903 |
-0.632 |
0.000 |
| C3 |
2.374 |
-0.230 |
0.000 |
| Cl4 |
-1.741 |
0.126 |
0.000 |
| H5 |
0.153 |
1.198 |
0.885 |
| H6 |
0.153 |
1.198 |
-0.885 |
| H7 |
0.678 |
-1.243 |
-0.877 |
| H8 |
0.678 |
-1.243 |
0.877 |
| H9 |
3.016 |
-1.112 |
0.000 |
| H10 |
2.626 |
0.363 |
-0.882 |
| H11 |
2.626 |
0.363 |
0.882 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 1.5138 | 2.5095 | 1.7998 | 1.0889 | 1.0889 | 2.1368 | 2.1368 | 3.4597 | 2.7790 | 2.7790 |
C2 | 1.5138 | | 1.5251 | 2.7501 | 2.1670 | 2.1670 | 1.0923 | 1.0923 | 2.1669 | 2.1764 | 2.1764 | C3 | 2.5095 | 1.5251 | | 4.1302 | 2.7852 | 2.7852 | 2.1613 | 2.1613 | 1.0909 | 1.0923 | 1.0923 | Cl4 | 1.7998 | 2.7501 | 4.1302 | | 2.3495 | 2.3495 | 2.9149 | 2.9149 | 4.9151 | 4.4612 | 4.4612 | H5 | 1.0889 | 2.1670 | 2.7852 | 2.3495 | | 1.7710 | 3.0568 | 2.4979 | 3.7840 | 3.1527 | 2.6104 | H6 | 1.0889 | 2.1670 | 2.7852 | 2.3495 | 1.7710 | | 2.4979 | 3.0568 | 3.7840 | 2.6104 | 3.1527 | H7 | 2.1368 | 1.0923 | 2.1613 | 2.9149 | 3.0568 | 2.4979 | | 1.7531 | 2.4998 | 2.5245 | 3.0768 | H8 | 2.1368 | 1.0923 | 2.1613 | 2.9149 | 2.4979 | 3.0568 | 1.7531 | | 2.4998 | 3.0768 | 2.5245 | H9 | 3.4597 | 2.1669 | 1.0909 | 4.9151 | 3.7840 | 3.7840 | 2.4998 | 2.4998 | | 1.7623 | 1.7623 | H10 | 2.7790 | 2.1764 | 1.0923 | 4.4612 | 3.1527 | 2.6104 | 2.5245 | 3.0768 | 1.7623 | | 1.7647 | H11 | 2.7790 | 2.1764 | 1.0923 | 4.4612 | 2.6104 | 3.1527 | 3.0768 | 2.5245 | 1.7623 | 1.7647 | |
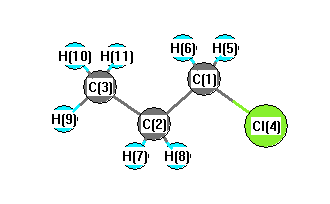 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
111.334 |
|
C1 |
C2 |
H7 |
109.088 |
| C1 |
C2 |
H8 |
109.088 |
|
C2 |
C1 |
Cl4 |
111.896 |
| C2 |
C1 |
H5 |
111.692 |
|
C2 |
C1 |
H6 |
111.692 |
| C2 |
C3 |
H9 |
110.754 |
|
C2 |
C3 |
H10 |
111.433 |
| C2 |
C3 |
H11 |
111.433 |
|
C3 |
C2 |
H7 |
110.234 |
| C3 |
C2 |
H8 |
110.234 |
|
Cl4 |
C1 |
H5 |
106.220 |
| Cl4 |
C1 |
H6 |
106.220 |
|
H5 |
C1 |
H6 |
108.814 |
| H7 |
C2 |
H8 |
106.741 |
|
H9 |
C3 |
H10 |
107.641 |
| H9 |
C3 |
H11 |
107.641 |
|
H10 |
C3 |
H11 |
107.760 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.091 |
|
|
|
| 2 |
C |
-0.153 |
|
|
|
| 3 |
C |
-0.282 |
|
|
|
| 4 |
Cl |
-0.187 |
|
|
|
| 5 |
H |
0.115 |
|
|
|
| 6 |
H |
0.115 |
|
|
|
| 7 |
H |
0.102 |
|
|
|
| 8 |
H |
0.102 |
|
|
|
| 9 |
H |
0.100 |
|
|
|
| 10 |
H |
0.090 |
|
|
|
| 11 |
H |
0.090 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.216 |
0.264 |
0.000 |
2.231 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.484 |
0.126 |
0.000 |
| y |
0.126 |
-32.444 |
0.000 |
| z |
0.000 |
0.000 |
-32.530 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.997 |
0.126 |
0.000 |
| y |
0.126 |
1.563 |
0.000 |
| z |
0.000 |
0.000 |
1.434 |
|
| Polar |
|---|
| 3z2-r2 | 2.869 |
|---|
| x2-y2 | -3.040 |
|---|
| xy | 0.126 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.462 |
-0.015 |
0.000 |
| y |
-0.015 |
6.503 |
0.000 |
| z |
0.000 |
0.000 |
6.051 |
<r2> (average value of r
2) Å
2
| <r2> |
152.141 |
| (<r2>)1/2 |
12.335 |
Jump to
S1C1
Energy calculated at B3PW91/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -578.725332 |
| Energy at 298.15K | -578.733080 |
| Nuclear repulsion energy | 162.524848 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3PW91/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3140 |
3019 |
11.56 |
|
|
|
| 2 |
A |
3122 |
3002 |
19.54 |
|
|
|
| 3 |
A |
3108 |
2989 |
34.32 |
|
|
|
| 4 |
A |
3085 |
2966 |
21.70 |
|
|
|
| 5 |
A |
3068 |
2950 |
4.47 |
|
|
|
| 6 |
A |
3039 |
2922 |
20.46 |
|
|
|
| 7 |
A |
3025 |
2909 |
21.34 |
|
|
|
| 8 |
A |
1500 |
1442 |
6.06 |
|
|
|
| 9 |
A |
1493 |
1436 |
8.32 |
|
|
|
| 10 |
A |
1476 |
1419 |
4.02 |
|
|
|
| 11 |
A |
1469 |
1413 |
3.92 |
|
|
|
| 12 |
A |
1409 |
1355 |
6.81 |
|
|
|
| 13 |
A |
1376 |
1323 |
0.28 |
|
|
|
| 14 |
A |
1328 |
1277 |
24.43 |
|
|
|
| 15 |
A |
1279 |
1230 |
3.88 |
|
|
|
| 16 |
A |
1235 |
1188 |
0.46 |
|
|
|
| 17 |
A |
1109 |
1066 |
0.64 |
|
|
|
| 18 |
A |
1082 |
1040 |
1.06 |
|
|
|
| 19 |
A |
1050 |
1009 |
3.07 |
|
|
|
| 20 |
A |
904 |
869 |
8.57 |
|
|
|
| 21 |
A |
866 |
833 |
4.99 |
|
|
|
| 22 |
A |
792 |
762 |
18.55 |
|
|
|
| 23 |
A |
663 |
637 |
17.80 |
|
|
|
| 24 |
A |
421 |
405 |
1.45 |
|
|
|
| 25 |
A |
293 |
281 |
0.73 |
|
|
|
| 26 |
A |
211 |
202 |
1.06 |
|
|
|
| 27 |
A |
135 |
129 |
0.88 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20837.5 cm
-1
Scaled (by 0.9616) Zero Point Vibrational Energy (zpe) 20037.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3PW91/cc-pVTZ
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.181 |
0.878 |
0.308 |
| C2 |
-1.143 |
0.561 |
-0.356 |
| C3 |
-1.806 |
-0.714 |
0.144 |
| Cl4 |
1.451 |
-0.346 |
-0.068 |
| H5 |
0.580 |
1.831 |
-0.030 |
| H6 |
0.094 |
0.894 |
1.394 |
| H7 |
-1.800 |
1.418 |
-0.168 |
| H8 |
-0.996 |
0.514 |
-1.437 |
| H9 |
-2.762 |
-0.876 |
-0.356 |
| H10 |
-1.175 |
-1.583 |
-0.046 |
| H11 |
-1.995 |
-0.665 |
1.219 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 1.5145 | 2.5510 | 1.8035 | 1.0879 | 1.0892 | 2.1076 | 2.1364 | 3.4897 | 2.8321 | 2.8184 |
C2 | 1.5145 | | 1.5215 | 2.7632 | 2.1657 | 2.1687 | 1.0959 | 1.0928 | 2.1650 | 2.1668 | 2.1699 | C3 | 2.5510 | 1.5215 | | 3.2845 | 3.4934 | 2.7858 | 2.1550 | 2.1595 | 1.0910 | 1.0904 | 1.0926 | Cl4 | 1.8035 | 2.7632 | 3.2845 | | 2.3450 | 2.3488 | 3.7001 | 2.9328 | 4.2562 | 2.9033 | 3.6922 | H5 | 1.0879 | 2.1657 | 3.4934 | 2.3450 | | 1.7726 | 2.4199 | 2.4902 | 4.3139 | 3.8395 | 3.7981 | H6 | 1.0892 | 2.1687 | 2.7858 | 2.3488 | 1.7726 | | 2.5105 | 3.0577 | 3.7888 | 3.1344 | 2.6130 | H7 | 2.1076 | 1.0959 | 2.1550 | 3.7001 | 2.4199 | 2.5105 | | 1.7537 | 2.4951 | 3.0681 | 2.5106 | H8 | 2.1364 | 1.0928 | 2.1595 | 2.9328 | 2.4902 | 3.0577 | 1.7537 | | 2.4944 | 2.5228 | 3.0730 | H9 | 3.4897 | 2.1650 | 1.0910 | 4.2562 | 4.3139 | 3.7888 | 2.4951 | 2.4944 | | 1.7646 | 1.7641 | H10 | 2.8321 | 2.1668 | 1.0904 | 2.9033 | 3.8395 | 3.1344 | 3.0681 | 2.5228 | 1.7646 | | 1.7648 | H11 | 2.8184 | 2.1699 | 1.0926 | 3.6922 | 3.7981 | 2.6130 | 2.5106 | 3.0730 | 1.7641 | 1.7648 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
114.333 |
|
C1 |
C2 |
H7 |
106.584 |
| C1 |
C2 |
H8 |
108.977 |
|
C2 |
C1 |
Cl4 |
112.483 |
| C2 |
C1 |
H5 |
111.597 |
|
C2 |
C1 |
H6 |
111.758 |
| C2 |
C3 |
H9 |
110.858 |
|
C2 |
C3 |
H10 |
111.041 |
| C2 |
C3 |
H11 |
111.154 |
|
C3 |
C2 |
H7 |
109.779 |
| C3 |
C2 |
H8 |
110.317 |
|
Cl4 |
C1 |
H5 |
105.715 |
| Cl4 |
C1 |
H6 |
105.925 |
|
H5 |
C1 |
H6 |
109.018 |
| H7 |
C2 |
H8 |
106.503 |
|
H9 |
C3 |
H10 |
107.984 |
| H9 |
C3 |
H11 |
107.778 |
|
H10 |
C3 |
H11 |
107.882 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.102 |
|
|
|
| 2 |
C |
-0.161 |
|
|
|
| 3 |
C |
-0.271 |
|
|
|
| 4 |
Cl |
-0.186 |
|
|
|
| 5 |
H |
0.124 |
|
|
|
| 6 |
H |
0.115 |
|
|
|
| 7 |
H |
0.091 |
|
|
|
| 8 |
H |
0.102 |
|
|
|
| 9 |
H |
0.095 |
|
|
|
| 10 |
H |
0.107 |
|
|
|
| 11 |
H |
0.084 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.679 |
1.275 |
0.290 |
2.128 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-34.704 |
0.239 |
0.136 |
| y |
0.239 |
-31.813 |
0.432 |
| z |
0.136 |
0.432 |
-32.621 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.487 |
0.239 |
0.136 |
| y |
0.239 |
1.850 |
0.432 |
| z |
0.136 |
0.432 |
0.637 |
|
| Polar |
|---|
| 3z2-r2 | 1.274 |
|---|
| x2-y2 | -2.892 |
|---|
| xy | 0.239 |
|---|
| xz | 0.136 |
|---|
| yz | 0.432 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.369 |
-0.453 |
-0.087 |
| y |
-0.453 |
7.175 |
0.118 |
| z |
-0.087 |
0.118 |
6.232 |
<r2> (average value of r
2) Å
2
| <r2> |
130.540 |
| (<r2>)1/2 |
11.425 |