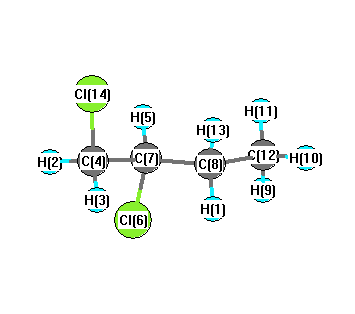
Vibrational Frequencies calculated at B3PW91/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3179 |
3061 |
4.36 |
|
|
|
| 2 |
A |
3130 |
3013 |
24.49 |
|
|
|
| 3 |
A |
3118 |
3002 |
44.75 |
|
|
|
| 4 |
A |
3113 |
2997 |
7.95 |
|
|
|
| 5 |
A |
3106 |
2990 |
1.01 |
|
|
|
| 6 |
A |
3082 |
2967 |
2.52 |
|
|
|
| 7 |
A |
3046 |
2933 |
34.84 |
|
|
|
| 8 |
A |
3045 |
2931 |
5.56 |
|
|
|
| 9 |
A |
1519 |
1463 |
10.48 |
|
|
|
| 10 |
A |
1512 |
1456 |
10.11 |
|
|
|
| 11 |
A |
1490 |
1434 |
4.77 |
|
|
|
| 12 |
A |
1484 |
1428 |
6.87 |
|
|
|
| 13 |
A |
1426 |
1373 |
5.19 |
|
|
|
| 14 |
A |
1410 |
1357 |
0.51 |
|
|
|
| 15 |
A |
1351 |
1301 |
4.66 |
|
|
|
| 16 |
A |
1340 |
1290 |
4.64 |
|
|
|
| 17 |
A |
1315 |
1266 |
10.02 |
|
|
|
| 18 |
A |
1260 |
1213 |
12.42 |
|
|
|
| 19 |
A |
1221 |
1175 |
11.77 |
|
|
|
| 20 |
A |
1139 |
1097 |
1.07 |
|
|
|
| 21 |
A |
1112 |
1070 |
2.04 |
|
|
|
| 22 |
A |
1076 |
1036 |
4.32 |
|
|
|
| 23 |
A |
1047 |
1008 |
0.80 |
|
|
|
| 24 |
A |
958 |
923 |
11.93 |
|
|
|
| 25 |
A |
838 |
807 |
6.92 |
|
|
|
| 26 |
A |
820 |
789 |
10.97 |
|
|
|
| 27 |
A |
742 |
714 |
55.51 |
|
|
|
| 28 |
A |
663 |
638 |
46.22 |
|
|
|
| 29 |
A |
462 |
445 |
2.00 |
|
|
|
| 30 |
A |
385 |
370 |
1.74 |
|
|
|
| 31 |
A |
295 |
284 |
0.06 |
|
|
|
| 32 |
A |
244 |
234 |
0.36 |
|
|
|
| 33 |
A |
219 |
210 |
2.35 |
|
|
|
| 34 |
A |
188 |
181 |
3.67 |
|
|
|
| 35 |
A |
115 |
111 |
3.80 |
|
|
|
| 36 |
A |
91 |
88 |
1.27 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 25269.7 cm
-1
Scaled (by 0.9627) Zero Point Vibrational Energy (zpe) 24327.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3PW91/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.237 |
|
|
|
| 2 |
H |
0.302 |
|
|
|
| 3 |
H |
0.284 |
|
|
|
| 4 |
C |
-0.489 |
|
|
|
| 5 |
H |
0.290 |
|
|
|
| 6 |
Cl |
-0.076 |
|
|
|
| 7 |
C |
-0.362 |
|
|
|
| 8 |
C |
-0.415 |
|
|
|
| 9 |
H |
0.248 |
|
|
|
| 10 |
H |
0.231 |
|
|
|
| 11 |
H |
0.223 |
|
|
|
| 12 |
C |
-0.652 |
|
|
|
| 13 |
H |
0.257 |
|
|
|
| 14 |
Cl |
-0.079 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.371 |
0.754 |
0.297 |
0.891 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-58.467 |
3.454 |
-0.218 |
| y |
3.454 |
-51.549 |
-0.206 |
| z |
-0.218 |
-0.206 |
-51.070 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-7.158 |
3.454 |
-0.218 |
| y |
3.454 |
3.220 |
-0.206 |
| z |
-0.218 |
-0.206 |
3.938 |
|
| Polar |
|---|
| 3z2-r2 | 7.875 |
|---|
| x2-y2 | -6.919 |
|---|
| xy | 3.454 |
|---|
| xz | -0.218 |
|---|
| yz | -0.206 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
323.477 |
| (<r2>)1/2 |
17.985 |