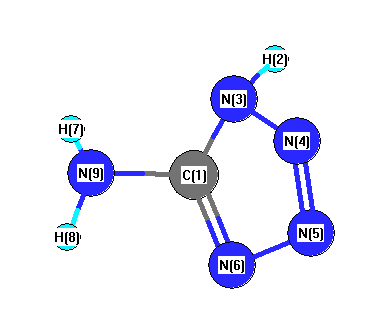
Vibrational Frequencies calculated at MP2=FULL/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3663 |
3451 |
17.87 |
|
|
|
| 2 |
A |
3663 |
3451 |
124.47 |
|
|
|
| 3 |
A |
3558 |
3352 |
31.94 |
|
|
|
| 4 |
A |
1712 |
1613 |
119.36 |
|
|
|
| 5 |
A |
1645 |
1550 |
115.97 |
|
|
|
| 6 |
A |
1531 |
1442 |
26.76 |
|
|
|
| 7 |
A |
1387 |
1307 |
0.68 |
|
|
|
| 8 |
A |
1208 |
1138 |
6.04 |
|
|
|
| 9 |
A |
1180 |
1111 |
16.92 |
|
|
|
| 10 |
A |
1141 |
1075 |
6.67 |
|
|
|
| 11 |
A |
1118 |
1053 |
16.55 |
|
|
|
| 12 |
A |
1077 |
1015 |
19.88 |
|
|
|
| 13 |
A |
1017 |
958 |
0.44 |
|
|
|
| 14 |
A |
827 |
779 |
237.48 |
|
|
|
| 15 |
A |
735 |
693 |
3.79 |
|
|
|
| 16 |
A |
722 |
680 |
8.68 |
|
|
|
| 17 |
A |
692 |
652 |
85.26 |
|
|
|
| 18 |
A |
562 |
530 |
78.47 |
|
|
|
| 19 |
A |
387 |
365 |
15.02 |
|
|
|
| 20 |
A |
302 |
284 |
1.86 |
|
|
|
| 21 |
A |
218 |
205 |
60.40 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14170.2 cm
-1
Scaled (by 0.9422) Zero Point Vibrational Energy (zpe) 13351.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.