Jump to
S1C2
S1C3
Energy calculated at MP2=FULL/cc-pVQZ
| | hartrees |
|---|
| Energy at 0K | -100.257624 |
| Energy at 298.15K | -100.256704 |
| HF Energy | -99.814530 |
| Nuclear repulsion energy | 27.536957 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP2=FULL/cc-pVQZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
-2.062 |
| C2 |
0.000 |
0.000 |
-0.156 |
| N3 |
0.000 |
0.000 |
1.018 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 1.9052 | 3.0794 |
C2 | 1.9052 | | 1.1742 | N3 | 3.0794 | 1.1742 | |
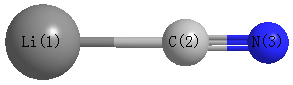 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
180.000 |
|
Li1 |
N3 |
C2 |
0.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at MP2=FULL/cc-pVQZ
| | hartrees |
|---|
| Energy at 0K | -100.259557 |
| Energy at 298.15K | -100.258860 |
| HF Energy | -99.820610 |
| Nuclear repulsion energy | 29.323075 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP2=FULL/cc-pVQZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
1.426 |
-0.593 |
0.000 |
| C2 |
-0.713 |
-0.371 |
0.000 |
| N3 |
0.000 |
0.572 |
0.000 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.1512 | 1.8415 |
C2 | 2.1512 | | 1.1818 | N3 | 1.8415 | 1.1818 | |
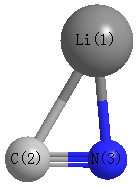 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
58.812 |
|
Li1 |
N3 |
C2 |
87.889 |
| C2 |
Li1 |
N3 |
33.299 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at MP2=FULL/cc-pVQZ
| | hartrees |
|---|
| Energy at 0K | -100.258774 |
| Energy at 298.15K | -100.257693 |
| HF Energy | -99.826026 |
| Nuclear repulsion energy | 28.343651 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP2=FULL/cc-pVQZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
1.880 |
| C2 |
0.000 |
0.000 |
-1.069 |
| N3 |
0.000 |
0.000 |
0.111 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.9498 | 1.7699 |
C2 | 2.9498 | | 1.1800 | N3 | 1.7699 | 1.1800 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
0.000 |
|
Li1 |
N3 |
C2 |
180.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability