Jump to
S1C2
S1C3
S1C4
Energy calculated at MP2=FULL/6-31G**
| | hartrees |
|---|
| Energy at 0K | -579.028342 |
| Energy at 298.15K | |
| HF Energy | -578.825443 |
| Nuclear repulsion energy | 66.793676 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2432 |
2273 |
0.00 |
279.06 |
0.35 |
0.52 |
| 2 |
Σg |
735 |
687 |
0.00 |
44.52 |
0.19 |
0.32 |
| 3 |
Σu |
2429 |
2269 |
0.61 |
0.00 |
0.00 |
0.00 |
| 4 |
Πg |
600i |
561i |
0.00 |
52.43 |
0.75 |
0.86 |
| 4 |
Πg |
600i |
561i |
0.00 |
52.43 |
0.75 |
0.86 |
| 5 |
Πu |
414 |
387 |
1.98 |
0.00 |
0.00 |
0.00 |
| 5 |
Πu |
414 |
387 |
1.98 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 2611.4 cm
-1
Scaled (by 0.9344) Zero Point Vibrational Energy (zpe) 2440.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/6-31G**
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
0.994 |
| Si2 |
0.000 |
0.000 |
-0.994 |
| H3 |
0.000 |
0.000 |
2.447 |
| H4 |
0.000 |
0.000 |
-2.447 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 1.9875 | 1.4534 | 3.4410 |
Si2 | 1.9875 | | 3.4410 | 1.4534 | H3 | 1.4534 | 3.4410 | | 4.8944 | H4 | 3.4410 | 1.4534 | 4.8944 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
180.000 |
|
Si2 |
Si1 |
H3 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
S1C4
Energy calculated at MP2=FULL/6-31G**
| | hartrees |
|---|
| Energy at 0K | -579.061932 |
| Energy at 298.15K | |
| HF Energy | -578.852574 |
| Nuclear repulsion energy | 64.091666 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2296 |
2145 |
0.00 |
437.85 |
0.27 |
0.42 |
| 2 |
Ag |
637 |
595 |
0.00 |
1846.38 |
0.33 |
0.50 |
| 3 |
Ag |
607 |
567 |
0.00 |
4029.73 |
0.39 |
0.56 |
| 4 |
Au |
979 |
914 |
850.91 |
0.00 |
0.00 |
0.00 |
| 5 |
Bu |
2304 |
2152 |
156.57 |
0.00 |
0.00 |
0.00 |
| 6 |
Bu |
498 |
465 |
37.64 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 3660.0 cm
-1
Scaled (by 0.9344) Zero Point Vibrational Energy (zpe) 3419.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/6-31G**
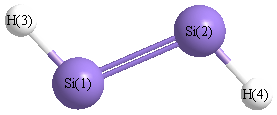
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.052 |
0.000 |
| Si2 |
0.000 |
-1.052 |
0.000 |
| H3 |
1.222 |
1.887 |
0.000 |
| H4 |
-1.222 |
-1.887 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.1034 | 1.4805 | 3.1828 |
Si2 | 2.1034 | | 3.1828 | 1.4805 | H3 | 1.4805 | 3.1828 | | 4.4967 | H4 | 3.1828 | 1.4805 | 4.4967 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
124.346 |
|
Si2 |
Si1 |
H3 |
124.346 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
S1C4
Energy calculated at MP2=FULL/6-31G**
| | hartrees |
|---|
| Energy at 0K | -579.087553 |
| Energy at 298.15K | |
| HF Energy | -578.890676 |
| Nuclear repulsion energy | 65.216823 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
1745 |
1631 |
4.59 |
59.54 |
0.07 |
0.13 |
| 2 |
A1 |
985 |
921 |
49.61 |
2.46 |
0.36 |
0.53 |
| 3 |
A1 |
543 |
507 |
1.78 |
52.63 |
0.35 |
0.52 |
| 4 |
A2 |
1262 |
1179 |
0.00 |
5.29 |
0.75 |
0.86 |
| 5 |
B1 |
1654 |
1546 |
21.63 |
17.10 |
0.75 |
0.86 |
| 6 |
B2 |
1314 |
1228 |
443.49 |
2.40 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 3751.5 cm
-1
Scaled (by 0.9344) Zero Point Vibrational Energy (zpe) 3505.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/6-31G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.102 |
-0.050 |
| Si2 |
0.000 |
-1.102 |
-0.050 |
| H3 |
0.979 |
0.000 |
0.702 |
| H4 |
-0.979 |
0.000 |
0.702 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.2049 | 1.6549 | 1.6549 |
Si2 | 2.2049 | | 1.6549 | 1.6549 | H3 | 1.6549 | 1.6549 | | 1.9575 | H4 | 1.6549 | 1.6549 | 1.9575 | |
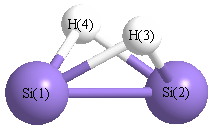 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
48.227 |
|
Si2 |
Si1 |
H3 |
48.227 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
S1C3
Energy calculated at MP2=FULL/6-31G**
| | hartrees |
|---|
| Energy at 0K | -579.072192 |
| Energy at 298.15K | |
| HF Energy | -578.869467 |
| Nuclear repulsion energy | 65.134571 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
2289 |
2139 |
114.07 |
278.30 |
0.41 |
0.58 |
| 2 |
A' |
1760 |
1645 |
93.45 |
82.91 |
0.23 |
0.38 |
| 3 |
A' |
1240 |
1159 |
242.75 |
4.43 |
0.68 |
0.81 |
| 4 |
A' |
620 |
579 |
15.24 |
20.88 |
0.37 |
0.54 |
| 5 |
A' |
482 |
450 |
6.12 |
1.09 |
0.15 |
0.26 |
| 6 |
A" |
230 |
215 |
32.74 |
2.58 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 3310.3 cm
-1
Scaled (by 0.9344) Zero Point Vibrational Energy (zpe) 3093.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/6-31G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.061 |
-1.144 |
0.000 |
| Si2 |
0.061 |
0.977 |
0.000 |
| H3 |
-1.220 |
-0.020 |
0.000 |
| H4 |
-0.482 |
2.353 |
0.000 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
| Si1 | | 2.1207 | 1.7033 | 3.5389 |
Si2 | 2.1207 | | 1.6231 | 1.4795 | H3 | 1.7033 | 1.6231 | | 2.4855 | H4 | 3.5389 | 1.4795 | 2.4855 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H4 |
158.465 |
|
Si2 |
Si1 |
H3 |
48.739 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability