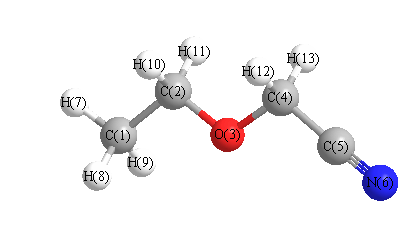
Vibrational Frequencies calculated at MP2=FULL/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3209 |
3050 |
14.37 |
|
|
|
| 2 |
A' |
3104 |
2950 |
12.30 |
|
|
|
| 3 |
A' |
3038 |
2888 |
43.59 |
|
|
|
| 4 |
A' |
3028 |
2878 |
31.35 |
|
|
|
| 5 |
A' |
2205 |
2096 |
9.99 |
|
|
|
| 6 |
A' |
1533 |
1457 |
2.92 |
|
|
|
| 7 |
A' |
1504 |
1430 |
1.10 |
|
|
|
| 8 |
A' |
1501 |
1427 |
4.64 |
|
|
|
| 9 |
A' |
1455 |
1383 |
9.89 |
|
|
|
| 10 |
A' |
1406 |
1336 |
27.88 |
|
|
|
| 11 |
A' |
1381 |
1312 |
54.86 |
|
|
|
| 12 |
A' |
1188 |
1129 |
190.35 |
|
|
|
| 13 |
A' |
1162 |
1104 |
15.07 |
|
|
|
| 14 |
A' |
1079 |
1026 |
20.51 |
|
|
|
| 15 |
A' |
967 |
919 |
6.17 |
|
|
|
| 16 |
A' |
916 |
870 |
5.85 |
|
|
|
| 17 |
A' |
549 |
522 |
0.69 |
|
|
|
| 18 |
A' |
426 |
405 |
0.86 |
|
|
|
| 19 |
A' |
296 |
281 |
1.57 |
|
|
|
| 20 |
A' |
130 |
124 |
2.82 |
|
|
|
| 21 |
A" |
3215 |
3056 |
14.74 |
|
|
|
| 22 |
A" |
3088 |
2934 |
4.36 |
|
|
|
| 23 |
A" |
3078 |
2925 |
75.13 |
|
|
|
| 24 |
A" |
1484 |
1410 |
6.31 |
|
|
|
| 25 |
A" |
1304 |
1240 |
2.56 |
|
|
|
| 26 |
A" |
1264 |
1201 |
3.85 |
|
|
|
| 27 |
A" |
1193 |
1134 |
7.12 |
|
|
|
| 28 |
A" |
1038 |
986 |
1.90 |
|
|
|
| 29 |
A" |
829 |
788 |
0.41 |
|
|
|
| 30 |
A" |
355 |
337 |
0.91 |
|
|
|
| 31 |
A" |
263 |
250 |
0.46 |
|
|
|
| 32 |
A" |
113 |
108 |
5.54 |
|
|
|
| 33 |
A" |
76 |
72 |
0.11 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23688.0 cm
-1
Scaled (by 0.9504) Zero Point Vibrational Energy (zpe) 22513.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.