Jump to
S1C2
S1C3
Vibrational Frequencies calculated at MP2=FULL/cc-pVDZ
Geometric Data calculated at MP2=FULL/cc-pVDZ
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Vibrational Frequencies calculated at MP2=FULL/cc-pVDZ
Geometric Data calculated at MP2=FULL/cc-pVDZ
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at MP2=FULL/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -267.599834 |
| Energy at 298.15K | -267.608205 |
| HF Energy | -266.808493 |
| Nuclear repulsion energy | 194.202698 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3195 |
3036 |
27.10 |
|
|
|
| 2 |
A |
3188 |
3030 |
38.16 |
|
|
|
| 3 |
A |
3155 |
2999 |
30.93 |
|
|
|
| 4 |
A |
3095 |
2941 |
55.74 |
|
|
|
| 5 |
A |
3061 |
2909 |
49.68 |
|
|
|
| 6 |
A |
2997 |
2848 |
116.92 |
|
|
|
| 7 |
A |
1548 |
1471 |
4.14 |
|
|
|
| 8 |
A |
1529 |
1453 |
2.23 |
|
|
|
| 9 |
A |
1511 |
1436 |
2.03 |
|
|
|
| 10 |
A |
1440 |
1369 |
13.81 |
|
|
|
| 11 |
A |
1383 |
1315 |
2.25 |
|
|
|
| 12 |
A |
1351 |
1284 |
0.61 |
|
|
|
| 13 |
A |
1286 |
1223 |
6.01 |
|
|
|
| 14 |
A |
1239 |
1178 |
0.99 |
|
|
|
| 15 |
A |
1225 |
1165 |
3.04 |
|
|
|
| 16 |
A |
1203 |
1143 |
82.61 |
|
|
|
| 17 |
A |
1164 |
1106 |
19.85 |
|
|
|
| 18 |
A |
1131 |
1075 |
101.21 |
|
|
|
| 19 |
A |
1095 |
1040 |
43.15 |
|
|
|
| 20 |
A |
1016 |
966 |
27.85 |
|
|
|
| 21 |
A |
987 |
938 |
45.79 |
|
|
|
| 22 |
A |
964 |
917 |
30.77 |
|
|
|
| 23 |
A |
873 |
830 |
10.34 |
|
|
|
| 24 |
A |
744 |
707 |
0.99 |
|
|
|
| 25 |
A |
692 |
657 |
5.17 |
|
|
|
| 26 |
A |
312 |
297 |
11.12 |
|
|
|
| 27 |
A |
59 |
56 |
3.03 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20720.6 cm
-1
Scaled (by 0.9504) Zero Point Vibrational Energy (zpe) 19692.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/cc-pVDZ
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
1.152 |
-0.109 |
0.172 |
| C2 |
-0.821 |
0.868 |
0.145 |
| C3 |
-1.042 |
-0.648 |
-0.037 |
| O4 |
0.519 |
1.056 |
-0.292 |
| O5 |
0.281 |
-1.189 |
-0.099 |
| H6 |
2.092 |
-0.252 |
-0.378 |
| H7 |
1.349 |
-0.035 |
1.264 |
| H8 |
-0.929 |
1.159 |
1.208 |
| H9 |
-1.561 |
-0.884 |
-0.980 |
| H10 |
-1.606 |
-1.087 |
0.803 |
| H11 |
-1.483 |
1.492 |
-0.471 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
O5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 2.2024 | 2.2692 | 1.4042 | 1.4139 | 1.0982 | 1.1126 | 2.6477 | 3.0475 | 2.9944 | 3.1493 |
C2 | 2.2024 | | 1.5427 | 1.4224 | 2.3461 | 3.1651 | 2.6035 | 1.1071 | 2.2090 | 2.2072 | 1.0980 | C3 | 2.2692 | 1.5427 | | 2.3251 | 1.4303 | 3.1773 | 2.7899 | 2.1966 | 1.1016 | 1.1032 | 2.2277 | O4 | 1.4042 | 1.4224 | 2.3251 | | 2.2656 | 2.0478 | 2.0735 | 2.0868 | 2.9263 | 3.2108 | 2.0567 | O5 | 1.4139 | 2.3461 | 1.4303 | 2.2656 | | 2.0580 | 2.0805 | 2.9460 | 2.0640 | 2.0940 | 3.2303 | H6 | 1.0982 | 3.1651 | 3.1773 | 2.0478 | 2.0580 | | 1.8159 | 3.6921 | 3.7555 | 3.9712 | 3.9788 | H7 | 1.1126 | 2.6035 | 2.7899 | 2.0735 | 2.0805 | 1.8159 | | 2.5719 | 3.7709 | 3.1702 | 3.6550 | H8 | 2.6477 | 1.1071 | 2.1966 | 2.0868 | 2.9460 | 3.6921 | 2.5719 | | 3.0586 | 2.3801 | 1.7986 | H9 | 3.0475 | 2.2090 | 1.1016 | 2.9263 | 2.0640 | 3.7555 | 3.7709 | 3.0586 | | 1.7950 | 2.4311 | H10 | 2.9944 | 2.2072 | 1.1032 | 3.2108 | 2.0940 | 3.9712 | 3.1702 | 2.3801 | 1.7950 | | 2.8793 | H11 | 3.1493 | 1.0980 | 2.2277 | 2.0567 | 3.2303 | 3.9788 | 3.6550 | 1.7986 | 2.4311 | 2.8793 | |
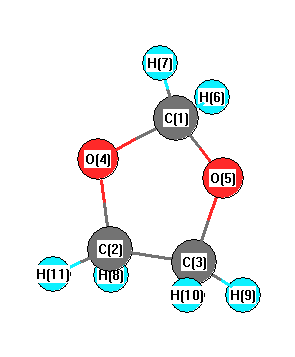 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O4 |
C2 |
102.364 |
|
C1 |
O5 |
C3 |
105.844 |
| C2 |
C3 |
O5 |
104.146 |
|
C2 |
C3 |
H9 |
112.241 |
| C2 |
C3 |
H10 |
111.994 |
|
C3 |
C2 |
O4 |
103.212 |
| C3 |
C2 |
H8 |
110.922 |
|
C3 |
C2 |
H11 |
113.988 |
| O4 |
C1 |
O5 |
107.015 |
|
O4 |
C1 |
H6 |
109.226 |
| O4 |
C1 |
H7 |
110.409 |
|
O4 |
C2 |
H8 |
110.556 |
| O4 |
C2 |
H11 |
108.694 |
|
O5 |
C1 |
H6 |
109.374 |
| O5 |
C1 |
H7 |
110.300 |
|
O5 |
C3 |
H9 |
108.520 |
| O5 |
C3 |
H10 |
110.825 |
|
H6 |
C1 |
H7 |
110.445 |
| H8 |
C2 |
H11 |
109.307 |
|
H9 |
C3 |
H10 |
109.002 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability