Jump to
S1C2
Vibrational Frequencies calculated at MP2=FULL/cc-pVDZ
Geometric Data calculated at MP2=FULL/cc-pVDZ
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2=FULL/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -623.935664 |
| Energy at 298.15K | -623.939443 |
| HF Energy | -623.211374 |
| Nuclear repulsion energy | 189.937941 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2=FULL/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3714 |
3530 |
101.10 |
|
|
|
| 2 |
A' |
1263 |
1200 |
145.27 |
|
|
|
| 3 |
A' |
1119 |
1063 |
15.44 |
|
|
|
| 4 |
A' |
759 |
721 |
161.69 |
|
|
|
| 5 |
A' |
471 |
447 |
25.76 |
|
|
|
| 6 |
A' |
434 |
413 |
135.50 |
|
|
|
| 7 |
A' |
319 |
303 |
16.21 |
|
|
|
| 8 |
A" |
3712 |
3528 |
34.72 |
|
|
|
| 9 |
A" |
1089 |
1035 |
52.00 |
|
|
|
| 10 |
A" |
753 |
715 |
333.54 |
|
|
|
| 11 |
A" |
435 |
413 |
57.40 |
|
|
|
| 12 |
A" |
173 |
164 |
17.58 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 7119.7 cm
-1
Scaled (by 0.9504) Zero Point Vibrational Energy (zpe) 6766.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2=FULL/cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.302 |
0.352 |
0.000 |
| O2 |
-1.048 |
0.971 |
0.000 |
| O3 |
0.302 |
-0.724 |
1.281 |
| O4 |
0.302 |
-0.724 |
-1.281 |
| H5 |
-0.645 |
-0.905 |
1.448 |
| H6 |
-0.645 |
-0.905 |
-1.448 |
Atom - Atom Distances (Å)
| |
S1 |
O2 |
O3 |
O4 |
H5 |
H6 |
| S1 | | 1.4852 | 1.6725 | 1.6725 | 2.1384 | 2.1384 |
O2 | 1.4852 | | 2.5169 | 2.5169 | 2.4039 | 2.4039 | O3 | 1.6725 | 2.5169 | | 2.5621 | 0.9783 | 2.8946 | O4 | 1.6725 | 2.5169 | 2.5621 | | 2.8946 | 0.9783 | H5 | 2.1384 | 2.4039 | 0.9783 | 2.8946 | | 2.8968 | H6 | 2.1384 | 2.4039 | 2.8946 | 0.9783 | 2.8968 | |
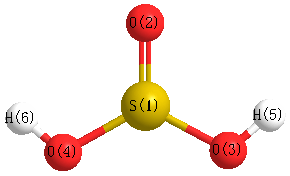 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
O3 |
H5 |
104.481 |
|
S1 |
O4 |
H6 |
104.481 |
| O2 |
S1 |
O3 |
105.549 |
|
O2 |
S1 |
O4 |
105.549 |
| O3 |
S1 |
O4 |
99.982 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability