Vibrational Frequencies calculated at mPW1PW91/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3164 |
3011 |
24.48 |
|
|
|
| 2 |
A |
3162 |
3009 |
13.47 |
|
|
|
| 3 |
A |
3148 |
2995 |
22.90 |
|
|
|
| 4 |
A |
3136 |
2984 |
30.00 |
|
|
|
| 5 |
A |
3125 |
2973 |
24.07 |
|
|
|
| 6 |
A |
3098 |
2948 |
28.02 |
|
|
|
| 7 |
A |
3083 |
2933 |
12.95 |
|
|
|
| 8 |
A |
3080 |
2930 |
23.53 |
|
|
|
| 9 |
A |
3069 |
2920 |
41.73 |
|
|
|
| 10 |
A |
3066 |
2917 |
7.66 |
|
|
|
| 11 |
A |
1520 |
1446 |
1.44 |
|
|
|
| 12 |
A |
1514 |
1440 |
6.14 |
|
|
|
| 13 |
A |
1504 |
1431 |
5.88 |
|
|
|
| 14 |
A |
1500 |
1427 |
4.35 |
|
|
|
| 15 |
A |
1496 |
1423 |
3.61 |
|
|
|
| 16 |
A |
1427 |
1357 |
4.46 |
|
|
|
| 17 |
A |
1394 |
1327 |
1.33 |
|
|
|
| 18 |
A |
1370 |
1303 |
0.40 |
|
|
|
| 19 |
A |
1330 |
1266 |
0.10 |
|
|
|
| 20 |
A |
1314 |
1251 |
4.79 |
|
|
|
| 21 |
A |
1297 |
1234 |
24.29 |
|
|
|
| 22 |
A |
1251 |
1190 |
4.73 |
|
|
|
| 23 |
A |
1215 |
1156 |
6.46 |
|
|
|
| 24 |
A |
1174 |
1117 |
0.58 |
|
|
|
| 25 |
A |
1123 |
1069 |
0.14 |
|
|
|
| 26 |
A |
1088 |
1035 |
2.27 |
|
|
|
| 27 |
A |
1053 |
1002 |
0.71 |
|
|
|
| 28 |
A |
1050 |
999 |
4.52 |
|
|
|
| 29 |
A |
982 |
934 |
3.32 |
|
|
|
| 30 |
A |
966 |
920 |
2.05 |
|
|
|
| 31 |
A |
917 |
873 |
0.89 |
|
|
|
| 32 |
A |
874 |
831 |
2.25 |
|
|
|
| 33 |
A |
862 |
820 |
2.14 |
|
|
|
| 34 |
A |
708 |
674 |
1.82 |
|
|
|
| 35 |
A |
649 |
618 |
2.81 |
|
|
|
| 36 |
A |
531 |
505 |
0.78 |
|
|
|
| 37 |
A |
464 |
442 |
0.08 |
|
|
|
| 38 |
A |
414 |
394 |
0.72 |
|
|
|
| 39 |
A |
286 |
272 |
1.11 |
|
|
|
| 40 |
A |
259 |
246 |
0.18 |
|
|
|
| 41 |
A |
244 |
232 |
0.20 |
|
|
|
| 42 |
A |
95 |
90 |
1.53 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 31498.7 cm
-1
Scaled (by 0.9515) Zero Point Vibrational Energy (zpe) 29971.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
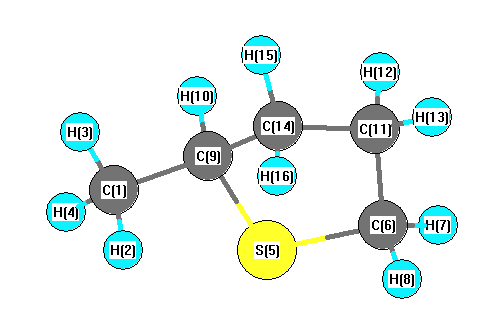
Charges from optimized geometry at mPW1PW91/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.385 |
|
|
|
| 2 |
H |
0.146 |
|
|
|
| 3 |
H |
0.133 |
|
|
|
| 4 |
H |
0.145 |
|
|
|
| 5 |
S |
0.085 |
|
|
|
| 6 |
C |
-0.403 |
|
|
|
| 7 |
H |
0.164 |
|
|
|
| 8 |
H |
0.163 |
|
|
|
| 9 |
C |
-0.274 |
|
|
|
| 10 |
H |
0.161 |
|
|
|
| 11 |
C |
-0.257 |
|
|
|
| 12 |
H |
0.144 |
|
|
|
| 13 |
H |
0.138 |
|
|
|
| 14 |
C |
-0.236 |
|
|
|
| 15 |
H |
0.137 |
|
|
|
| 16 |
H |
0.139 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.372 |
2.135 |
0.016 |
2.168 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-41.745 |
-0.333 |
0.961 |
| y |
-0.333 |
-47.771 |
-0.354 |
| z |
0.961 |
-0.354 |
-45.492 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.887 |
-0.333 |
0.961 |
| y |
-0.333 |
-4.152 |
-0.354 |
| z |
0.961 |
-0.354 |
-0.734 |
|
| Polar |
|---|
| 3z2-r2 | -1.469 |
|---|
| x2-y2 | 6.026 |
|---|
| xy | -0.333 |
|---|
| xz | 0.961 |
|---|
| yz | -0.354 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
203.021 |
| (<r2>)1/2 |
14.249 |