Vibrational Frequencies calculated at mPW1PW91/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3735 |
3555 |
77.69 |
|
|
|
| 2 |
A' |
3305 |
3146 |
2.33 |
|
|
|
| 3 |
A' |
3286 |
3127 |
1.03 |
|
|
|
| 4 |
A' |
3238 |
3082 |
16.55 |
|
|
|
| 5 |
A' |
3227 |
3071 |
28.12 |
|
|
|
| 6 |
A' |
3216 |
3061 |
2.98 |
|
|
|
| 7 |
A' |
3210 |
3055 |
1.41 |
|
|
|
| 8 |
A' |
1694 |
1612 |
2.82 |
|
|
|
| 9 |
A' |
1650 |
1571 |
1.05 |
|
|
|
| 10 |
A' |
1578 |
1502 |
10.64 |
|
|
|
| 11 |
A' |
1545 |
1471 |
4.61 |
|
|
|
| 12 |
A' |
1506 |
1433 |
24.82 |
|
|
|
| 13 |
A' |
1475 |
1404 |
13.58 |
|
|
|
| 14 |
A' |
1406 |
1338 |
38.89 |
|
|
|
| 15 |
A' |
1394 |
1327 |
5.22 |
|
|
|
| 16 |
A' |
1320 |
1257 |
9.89 |
|
|
|
| 17 |
A' |
1277 |
1216 |
9.97 |
|
|
|
| 18 |
A' |
1239 |
1179 |
4.24 |
|
|
|
| 19 |
A' |
1181 |
1124 |
1.10 |
|
|
|
| 20 |
A' |
1154 |
1098 |
0.72 |
|
|
|
| 21 |
A' |
1125 |
1071 |
29.50 |
|
|
|
| 22 |
A' |
1099 |
1046 |
5.42 |
|
|
|
| 23 |
A' |
1047 |
997 |
7.72 |
|
|
|
| 24 |
A' |
914 |
870 |
6.46 |
|
|
|
| 25 |
A' |
888 |
846 |
0.49 |
|
|
|
| 26 |
A' |
782 |
744 |
2.72 |
|
|
|
| 27 |
A' |
618 |
588 |
1.43 |
|
|
|
| 28 |
A' |
553 |
526 |
0.06 |
|
|
|
| 29 |
A' |
405 |
386 |
4.66 |
|
|
|
| 30 |
A" |
987 |
940 |
0.03 |
|
|
|
| 31 |
A" |
948 |
902 |
2.30 |
|
|
|
| 32 |
A" |
877 |
834 |
2.80 |
|
|
|
| 33 |
A" |
864 |
822 |
0.25 |
|
|
|
| 34 |
A" |
789 |
751 |
12.59 |
|
|
|
| 35 |
A" |
758 |
721 |
119.76 |
|
|
|
| 36 |
A" |
734 |
699 |
28.77 |
|
|
|
| 37 |
A" |
620 |
590 |
8.08 |
|
|
|
| 38 |
A" |
588 |
559 |
0.82 |
|
|
|
| 39 |
A" |
434 |
413 |
1.38 |
|
|
|
| 40 |
A" |
412 |
392 |
77.67 |
|
|
|
| 41 |
A" |
247 |
235 |
0.15 |
|
|
|
| 42 |
A" |
216 |
206 |
12.47 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 28769.8 cm
-1
Scaled (by 0.9518) Zero Point Vibrational Energy (zpe) 27383.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
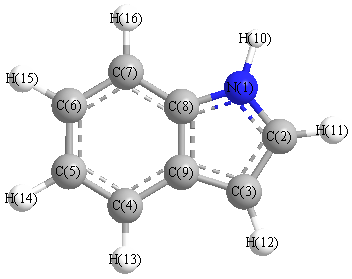
Charges from optimized geometry at mPW1PW91/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.259 |
|
|
|
| 2 |
C |
-0.129 |
|
|
|
| 3 |
C |
0.097 |
|
|
|
| 4 |
C |
-0.684 |
|
|
|
| 5 |
C |
0.072 |
|
|
|
| 6 |
C |
-0.476 |
|
|
|
| 7 |
C |
0.182 |
|
|
|
| 8 |
C |
-1.618 |
|
|
|
| 9 |
C |
1.553 |
|
|
|
| 10 |
H |
0.321 |
|
|
|
| 11 |
H |
0.163 |
|
|
|
| 12 |
H |
0.163 |
|
|
|
| 13 |
H |
0.158 |
|
|
|
| 14 |
H |
0.153 |
|
|
|
| 15 |
H |
0.154 |
|
|
|
| 16 |
H |
0.149 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.878 |
2.000 |
0.000 |
2.184 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.771 |
-2.111 |
0.000 |
| y |
-2.111 |
-41.932 |
0.000 |
| z |
0.000 |
0.000 |
-58.504 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.447 |
-2.111 |
0.000 |
| y |
-2.111 |
10.705 |
0.000 |
| z |
0.000 |
0.000 |
-14.153 |
|
| Polar |
|---|
| 3z2-r2 | -28.305 |
|---|
| x2-y2 | -4.839 |
|---|
| xy | -2.111 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
19.332 |
-1.552 |
0.000 |
| y |
-1.552 |
15.612 |
0.000 |
| z |
0.000 |
0.000 |
8.056 |
<r2> (average value of r
2) Å
2
| <r2> |
281.630 |
| (<r2>)1/2 |
16.782 |