Jump to
S1C2
S1C3
Vibrational Frequencies calculated at mPW1PW91/6-31+G**
Geometric Data calculated at mPW1PW91/6-31+G**
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at mPW1PW91/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -351.762816 |
| Energy at 298.15K | -351.764028 |
| HF Energy | -351.762816 |
| Nuclear repulsion energy | 81.761675 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
4083 |
3886 |
0.00 |
|
|
|
| 2 |
A |
592 |
563 |
0.00 |
|
|
|
| 3 |
A |
262 |
249 |
0.00 |
|
|
|
| 4 |
A |
161 |
153 |
28.45 |
|
|
|
| 5 |
A |
58 |
55 |
331.72 |
|
|
|
| 6 |
B |
4083 |
3886 |
168.31 |
|
|
|
| 7 |
B |
880 |
838 |
172.32 |
|
|
|
| 8 |
B |
252 |
240 |
344.42 |
|
|
|
| 9 |
B |
155 |
147 |
50.35 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 5262.4 cm
-1
Scaled (by 0.9518) Zero Point Vibrational Energy (zpe) 5008.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
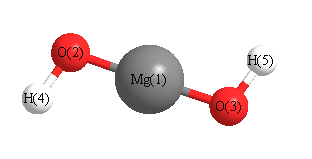
Geometric Data calculated at mPW1PW91/6-31+G**
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Mg1 |
0.000 |
0.000 |
0.000 |
| O2 |
0.000 |
1.795 |
-0.000 |
| O3 |
0.000 |
-1.795 |
-0.000 |
| H4 |
-0.507 |
2.599 |
0.000 |
| H5 |
0.507 |
-2.599 |
0.000 |
Atom - Atom Distances (Å)
| |
Mg1 |
O2 |
O3 |
H4 |
H5 |
| Mg1 | | 1.7947 | 1.7947 | 2.6480 | 2.6480 |
O2 | 1.7947 | | 3.5894 | 0.9509 | 4.4228 | O3 | 1.7947 | 3.5894 | | 4.4228 | 0.9509 | H4 | 2.6480 | 0.9509 | 4.4228 | | 5.2960 | H5 | 2.6480 | 4.4228 | 0.9509 | 5.2960 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Mg1 |
O2 |
H4 |
147.750 |
|
Mg1 |
O3 |
H5 |
147.750 |
| O2 |
Mg1 |
O3 |
179.999 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Mg |
1.187 |
|
|
|
| 2 |
O |
-0.953 |
|
|
|
| 3 |
O |
-0.953 |
|
|
|
| 4 |
H |
0.359 |
|
|
|
| 5 |
H |
0.359 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-18.922 |
-5.520 |
0.000 |
| y |
-5.520 |
-24.370 |
0.000 |
| z |
0.000 |
0.000 |
-20.524 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.526 |
-5.520 |
0.000 |
| y |
-5.520 |
-4.647 |
0.000 |
| z |
0.000 |
0.000 |
1.122 |
|
| Polar |
|---|
| 3z2-r2 | 2.243 |
|---|
| x2-y2 | 5.449 |
|---|
| xy | -5.520 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.129 |
0.316 |
0.000 |
| y |
0.316 |
4.648 |
0.000 |
| z |
0.000 |
0.000 |
3.285 |
<r2> (average value of r
2) Å
2
| <r2> |
78.845 |
| (<r2>)1/2 |
8.879 |
Jump to
S1C1
S1C2
Energy calculated at mPW1PW91/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -351.762235 |
| Energy at 298.15K | |
| HF Energy | -351.762235 |
| Nuclear repulsion energy | 82.389836 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
4130 |
3931 |
0.00 |
|
|
|
| 2 |
Σg |
602 |
573 |
0.00 |
|
|
|
| 3 |
Σu |
4129 |
3930 |
233.87 |
|
|
|
| 4 |
Σu |
901 |
857 |
216.27 |
|
|
|
| 5 |
Πg |
176i |
168i |
0.00 |
|
|
|
| 5 |
Πg |
176i |
168i |
0.00 |
|
|
|
| 6 |
Πu |
170 |
162 |
31.58 |
|
|
|
| 6 |
Πu |
170 |
162 |
31.58 |
|
|
|
| 7 |
Πu |
148i |
141i |
333.46 |
|
|
|
| 7 |
Πu |
148i |
141i |
333.46 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 4726.3 cm
-1
Scaled (by 0.9518) Zero Point Vibrational Energy (zpe) 4498.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at mPW1PW91/6-31+G**
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Mg1 |
0.000 |
0.000 |
0.000 |
| O2 |
0.000 |
0.000 |
1.774 |
| O3 |
0.000 |
0.000 |
-1.774 |
| H4 |
0.000 |
0.000 |
2.722 |
| H5 |
0.000 |
0.000 |
-2.722 |
Atom - Atom Distances (Å)
| |
Mg1 |
O2 |
O3 |
H4 |
H5 |
| Mg1 | | 1.7743 | 1.7743 | 2.7219 | 2.7219 |
O2 | 1.7743 | | 3.5485 | 0.9476 | 4.4961 | O3 | 1.7743 | 3.5485 | | 4.4961 | 0.9476 | H4 | 2.7219 | 0.9476 | 4.4961 | | 5.4438 | H5 | 2.7219 | 4.4961 | 0.9476 | 5.4438 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Mg1 |
O2 |
H4 |
180.000 |
|
Mg1 |
O3 |
H5 |
180.000 |
| O2 |
Mg1 |
O3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Mg |
1.274 |
|
|
|
| 2 |
O |
-0.996 |
|
|
|
| 3 |
O |
-0.996 |
|
|
|
| 4 |
H |
0.359 |
|
|
|
| 5 |
H |
0.359 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-20.447 |
0.000 |
0.000 |
| y |
0.000 |
-20.447 |
0.000 |
| z |
0.000 |
0.000 |
-20.567 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.060 |
0.000 |
0.000 |
| y |
0.000 |
0.060 |
0.000 |
| z |
0.000 |
0.000 |
-0.120 |
|
| Polar |
|---|
| 3z2-r2 | -0.239 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.106 |
0.000 |
0.000 |
| y |
0.000 |
3.106 |
0.000 |
| z |
0.000 |
0.000 |
4.411 |
<r2> (average value of r
2) Å
2
| <r2> |
77.981 |
| (<r2>)1/2 |
8.831 |