Jump to
S1C2
Energy calculated at mPW1PW91/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -358.394311 |
| Energy at 298.15K | -358.399409 |
| Nuclear repulsion energy | 232.675694 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3817 |
3633 |
95.77 |
|
|
|
| 2 |
A' |
3786 |
3603 |
95.74 |
|
|
|
| 3 |
A' |
3638 |
3462 |
68.69 |
|
|
|
| 4 |
A' |
1853 |
1763 |
26.68 |
|
|
|
| 5 |
A' |
1838 |
1749 |
668.22 |
|
|
|
| 6 |
A' |
1617 |
1539 |
120.13 |
|
|
|
| 7 |
A' |
1451 |
1381 |
27.91 |
|
|
|
| 8 |
A' |
1336 |
1272 |
91.88 |
|
|
|
| 9 |
A' |
1208 |
1149 |
255.71 |
|
|
|
| 10 |
A' |
1106 |
1053 |
3.46 |
|
|
|
| 11 |
A' |
792 |
754 |
9.93 |
|
|
|
| 12 |
A' |
614 |
585 |
74.83 |
|
|
|
| 13 |
A' |
533 |
507 |
0.21 |
|
|
|
| 14 |
A' |
417 |
397 |
4.23 |
|
|
|
| 15 |
A' |
269 |
256 |
16.06 |
|
|
|
| 16 |
A" |
836 |
796 |
6.29 |
|
|
|
| 17 |
A" |
679 |
647 |
138.13 |
|
|
|
| 18 |
A" |
633 |
603 |
23.90 |
|
|
|
| 19 |
A" |
445 |
424 |
32.25 |
|
|
|
| 20 |
A" |
373 |
355 |
205.62 |
|
|
|
| 21 |
A" |
68 |
65 |
5.70 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13653.4 cm
-1
Scaled (by 0.9518) Zero Point Vibrational Energy (zpe) 12995.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at mPW1PW91/6-31+G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.753 |
0.000 |
| C2 |
-0.056 |
-0.786 |
0.000 |
| O3 |
-1.101 |
-1.403 |
0.000 |
| O4 |
1.040 |
1.373 |
0.000 |
| O5 |
-1.207 |
1.302 |
0.000 |
| N6 |
1.184 |
-1.316 |
0.000 |
| H7 |
1.289 |
-2.316 |
0.000 |
| H8 |
1.990 |
-0.712 |
0.000 |
| H9 |
-1.088 |
2.263 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5403 | 2.4213 | 1.2108 | 1.3256 | 2.3836 | 3.3289 | 2.4715 | 1.8608 |
C2 | 1.5403 | | 1.2137 | 2.4211 | 2.3840 | 1.3481 | 2.0371 | 2.0477 | 3.2189 | O3 | 2.4213 | 1.2137 | | 3.5059 | 2.7071 | 2.2864 | 2.5583 | 3.1676 | 3.6664 | O4 | 1.2108 | 2.4211 | 3.5059 | | 2.2483 | 2.6921 | 3.6970 | 2.2909 | 2.3068 | O5 | 1.3256 | 2.3840 | 2.7071 | 2.2483 | | 3.5448 | 4.3952 | 3.7785 | 0.9687 | N6 | 2.3836 | 1.3481 | 2.2864 | 2.6921 | 3.5448 | | 1.0059 | 1.0074 | 4.2387 | H7 | 3.3289 | 2.0371 | 2.5583 | 3.6970 | 4.3952 | 1.0059 | | 1.7506 | 5.1592 | H8 | 2.4715 | 2.0477 | 3.1676 | 2.2909 | 3.7785 | 1.0074 | 1.7506 | | 4.2808 | H9 | 1.8608 | 3.2189 | 3.6664 | 2.3068 | 0.9687 | 4.2387 | 5.1592 | 4.2808 | |
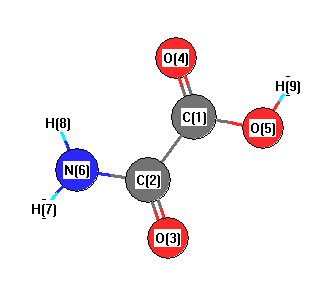 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
122.654 |
|
C1 |
C2 |
N6 |
111.049 |
| C1 |
O5 |
H9 |
107.378 |
|
C2 |
C1 |
O4 |
122.852 |
| C2 |
C1 |
O5 |
112.359 |
|
C2 |
N6 |
H7 |
119.138 |
| C2 |
N6 |
H8 |
120.056 |
|
O3 |
C2 |
N6 |
126.297 |
| O4 |
C1 |
O5 |
124.789 |
|
H7 |
N6 |
H8 |
120.806 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.431 |
|
|
|
| 2 |
C |
0.467 |
|
|
|
| 3 |
O |
-0.510 |
|
|
|
| 4 |
O |
-0.484 |
|
|
|
| 5 |
O |
-0.406 |
|
|
|
| 6 |
N |
-0.574 |
|
|
|
| 7 |
H |
0.346 |
|
|
|
| 8 |
H |
0.348 |
|
|
|
| 9 |
H |
0.383 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.261 |
0.696 |
0.000 |
2.366 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.727 |
8.331 |
0.009 |
| y |
8.331 |
-39.776 |
0.004 |
| z |
0.009 |
0.004 |
-34.029 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
12.175 |
8.331 |
0.009 |
| y |
8.331 |
-10.398 |
0.004 |
| z |
0.009 |
0.004 |
-1.777 |
|
| Polar |
|---|
| 3z2-r2 | -3.555 |
|---|
| x2-y2 | 15.049 |
|---|
| xy | 8.331 |
|---|
| xz | 0.009 |
|---|
| yz | 0.004 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
141.757 |
| (<r2>)1/2 |
11.906 |
Jump to
S1C1
Energy calculated at mPW1PW91/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -358.402564 |
| Energy at 298.15K | -358.407888 |
| Nuclear repulsion energy | 234.130790 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3775 |
3593 |
104.26 |
|
|
|
| 2 |
A' |
3640 |
3464 |
172.69 |
|
|
|
| 3 |
A' |
3629 |
3454 |
67.26 |
|
|
|
| 4 |
A' |
1894 |
1802 |
277.60 |
|
|
|
| 5 |
A' |
1823 |
1735 |
376.88 |
|
|
|
| 6 |
A' |
1621 |
1543 |
75.14 |
|
|
|
| 7 |
A' |
1464 |
1394 |
168.75 |
|
|
|
| 8 |
A' |
1359 |
1294 |
404.26 |
|
|
|
| 9 |
A' |
1232 |
1173 |
1.40 |
|
|
|
| 10 |
A' |
1110 |
1056 |
5.78 |
|
|
|
| 11 |
A' |
818 |
778 |
9.90 |
|
|
|
| 12 |
A' |
633 |
602 |
13.17 |
|
|
|
| 13 |
A' |
548 |
522 |
2.40 |
|
|
|
| 14 |
A' |
405 |
385 |
7.74 |
|
|
|
| 15 |
A' |
268 |
255 |
44.51 |
|
|
|
| 16 |
A" |
822 |
783 |
1.40 |
|
|
|
| 17 |
A" |
757 |
721 |
115.55 |
|
|
|
| 18 |
A" |
663 |
631 |
2.80 |
|
|
|
| 19 |
A" |
485 |
461 |
189.76 |
|
|
|
| 20 |
A" |
413 |
393 |
78.66 |
|
|
|
| 21 |
A" |
117 |
111 |
8.58 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13737.8 cm
-1
Scaled (by 0.9518) Zero Point Vibrational Energy (zpe) 13075.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at mPW1PW91/6-31+G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.011 |
-0.794 |
0.000 |
| C2 |
0.000 |
0.746 |
0.000 |
| O3 |
-1.075 |
1.333 |
0.000 |
| O4 |
1.024 |
-1.446 |
0.000 |
| O5 |
-1.215 |
-1.290 |
0.000 |
| N6 |
1.218 |
1.294 |
0.000 |
| H7 |
1.323 |
2.295 |
0.000 |
| H8 |
2.026 |
0.690 |
0.000 |
| H9 |
-1.820 |
-0.521 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5406 | 2.3883 | 1.2049 | 1.3224 | 2.4122 | 3.3563 | 2.5030 | 1.8509 |
C2 | 1.5406 | | 1.2247 | 2.4199 | 2.3714 | 1.3359 | 2.0369 | 2.0271 | 2.2176 | O3 | 2.3883 | 1.2247 | | 3.4828 | 2.6268 | 2.2939 | 2.5839 | 3.1672 | 1.9980 | O4 | 1.2049 | 2.4199 | 3.4828 | | 2.2448 | 2.7469 | 3.7529 | 2.3595 | 2.9907 | O5 | 1.3224 | 2.3714 | 2.6268 | 2.2448 | | 3.5496 | 4.3926 | 3.7984 | 0.9783 | N6 | 2.4122 | 1.3359 | 2.2939 | 2.7469 | 3.5496 | | 1.0065 | 1.0085 | 3.5391 | H7 | 3.3563 | 2.0369 | 2.5839 | 3.7529 | 4.3926 | 1.0065 | | 1.7521 | 4.2199 | H8 | 2.5030 | 2.0271 | 3.1672 | 2.3595 | 3.7984 | 1.0085 | 1.7521 | | 4.0322 | H9 | 1.8509 | 2.2176 | 1.9980 | 2.9907 | 0.9783 | 3.5391 | 4.2199 | 4.0322 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
119.026 |
|
C1 |
C2 |
N6 |
113.795 |
| C1 |
O5 |
H9 |
106.154 |
|
C2 |
C1 |
O4 |
123.153 |
| C2 |
C1 |
O5 |
111.618 |
|
C2 |
N6 |
H7 |
120.166 |
| C2 |
N6 |
H8 |
119.022 |
|
O3 |
C2 |
N6 |
127.179 |
| O4 |
C1 |
O5 |
125.229 |
|
H7 |
N6 |
H8 |
120.813 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.493 |
|
|
|
| 2 |
C |
0.431 |
|
|
|
| 3 |
O |
-0.562 |
|
|
|
| 4 |
O |
-0.478 |
|
|
|
| 5 |
O |
-0.408 |
|
|
|
| 6 |
N |
-0.564 |
|
|
|
| 7 |
H |
0.350 |
|
|
|
| 8 |
H |
0.354 |
|
|
|
| 9 |
H |
0.384 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.568 |
2.704 |
0.000 |
3.126 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.757 |
7.656 |
0.000 |
| y |
7.656 |
-38.100 |
0.000 |
| z |
0.000 |
0.000 |
-33.950 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.268 |
7.656 |
0.000 |
| y |
7.656 |
-5.746 |
0.000 |
| z |
0.000 |
0.000 |
0.478 |
|
| Polar |
|---|
| 3z2-r2 | 0.957 |
|---|
| x2-y2 | 7.343 |
|---|
| xy | 7.656 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.820 |
-0.115 |
0.000 |
| y |
-0.115 |
6.339 |
0.000 |
| z |
0.000 |
0.000 |
3.714 |
<r2> (average value of r
2) Å
2
| <r2> |
139.534 |
| (<r2>)1/2 |
11.812 |