Jump to
S1C2
Energy calculated at mPW1PW91/3-21G*
| | hartrees |
|---|
| Energy at 0K | -340.415214 |
| Energy at 298.15K | |
| HF Energy | -340.415214 |
| Nuclear repulsion energy | 241.390751 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3115 |
2958 |
33.19 |
|
|
|
| 2 |
A1 |
1943 |
1845 |
478.46 |
|
|
|
| 3 |
A1 |
1593 |
1513 |
1.24 |
|
|
|
| 4 |
A1 |
1386 |
1316 |
0.01 |
|
|
|
| 5 |
A1 |
1147 |
1089 |
187.67 |
|
|
|
| 6 |
A1 |
923 |
876 |
6.13 |
|
|
|
| 7 |
A1 |
787 |
748 |
30.19 |
|
|
|
| 8 |
A1 |
710 |
674 |
1.77 |
|
|
|
| 9 |
A2 |
3147 |
2989 |
0.00 |
|
|
|
| 10 |
A2 |
1278 |
1213 |
0.00 |
|
|
|
| 11 |
A2 |
1151 |
1093 |
0.00 |
|
|
|
| 12 |
A2 |
169i |
161i |
0.00 |
|
|
|
| 13 |
B1 |
3171 |
3012 |
34.14 |
|
|
|
| 14 |
B1 |
1266 |
1202 |
0.12 |
|
|
|
| 15 |
B1 |
877 |
833 |
0.24 |
|
|
|
| 16 |
B1 |
731 |
694 |
26.29 |
|
|
|
| 17 |
B1 |
123 |
117 |
4.33 |
|
|
|
| 18 |
B2 |
3107 |
2950 |
23.96 |
|
|
|
| 19 |
B2 |
1578 |
1499 |
2.54 |
|
|
|
| 20 |
B2 |
1416 |
1345 |
0.00 |
|
|
|
| 21 |
B2 |
1150 |
1092 |
6.66 |
|
|
|
| 22 |
B2 |
963 |
914 |
295.00 |
|
|
|
| 23 |
B2 |
740 |
703 |
1.37 |
|
|
|
| 24 |
B2 |
489 |
464 |
0.42 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16310.4 cm
-1
Scaled (by 0.9496) Zero Point Vibrational Energy (zpe) 15488.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at mPW1PW91/3-21G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.892 |
| O2 |
0.000 |
0.000 |
2.087 |
| O3 |
0.000 |
1.153 |
0.058 |
| O4 |
0.000 |
-1.153 |
0.058 |
| C5 |
0.000 |
0.783 |
-1.315 |
| C6 |
0.000 |
-0.783 |
-1.315 |
| H7 |
-0.892 |
1.184 |
-1.799 |
| H8 |
0.892 |
1.184 |
-1.799 |
| H9 |
0.892 |
-1.184 |
-1.799 |
| H10 |
-0.892 |
-1.184 |
-1.799 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1953 | 1.4228 | 1.4228 | 2.3416 | 2.3416 | 3.0720 | 3.0720 | 3.0720 | 3.0720 |
O2 | 1.1953 | | 2.3335 | 2.3335 | 3.4911 | 3.4911 | 4.1591 | 4.1591 | 4.1591 | 4.1591 | O3 | 1.4228 | 2.3335 | | 2.3068 | 1.4228 | 2.3739 | 2.0608 | 2.0608 | 3.1158 | 3.1158 | O4 | 1.4228 | 2.3335 | 2.3068 | | 2.3739 | 1.4228 | 3.1158 | 3.1158 | 2.0608 | 2.0608 | C5 | 2.3416 | 3.4911 | 1.4228 | 2.3739 | | 1.5655 | 1.0912 | 1.0912 | 2.2129 | 2.2129 | C6 | 2.3416 | 3.4911 | 2.3739 | 1.4228 | 1.5655 | | 2.2129 | 2.2129 | 1.0912 | 1.0912 | H7 | 3.0720 | 4.1591 | 2.0608 | 3.1158 | 1.0912 | 2.2129 | | 1.7843 | 2.9646 | 2.3676 | H8 | 3.0720 | 4.1591 | 2.0608 | 3.1158 | 1.0912 | 2.2129 | 1.7843 | | 2.3676 | 2.9646 | H9 | 3.0720 | 4.1591 | 3.1158 | 2.0608 | 2.2129 | 1.0912 | 2.9646 | 2.3676 | | 1.7843 | H10 | 3.0720 | 4.1591 | 3.1158 | 2.0608 | 2.2129 | 1.0912 | 2.3676 | 2.9646 | 1.7843 | |
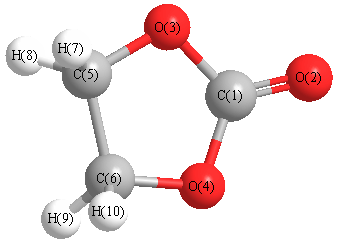 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
110.745 |
|
C1 |
O4 |
C6 |
110.745 |
| O2 |
C1 |
O3 |
125.844 |
|
O2 |
C1 |
O4 |
125.844 |
| O3 |
C1 |
O4 |
108.312 |
|
O3 |
C5 |
C6 |
105.099 |
| O3 |
C5 |
H7 |
109.407 |
|
O3 |
C5 |
H8 |
109.407 |
| O4 |
C6 |
C5 |
105.099 |
|
O4 |
C6 |
H9 |
109.407 |
| O4 |
C6 |
H10 |
109.407 |
|
C5 |
C6 |
H9 |
111.564 |
| C5 |
C6 |
H10 |
111.564 |
|
C6 |
C5 |
H7 |
111.564 |
| C6 |
C5 |
H8 |
111.564 |
|
H7 |
C5 |
H8 |
109.681 |
| H9 |
C6 |
H10 |
109.681 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.890 |
|
|
|
| 2 |
O |
-0.472 |
|
|
|
| 3 |
O |
-0.508 |
|
|
|
| 4 |
O |
-0.508 |
|
|
|
| 5 |
C |
-0.206 |
|
|
|
| 6 |
C |
-0.206 |
|
|
|
| 7 |
H |
0.252 |
|
|
|
| 8 |
H |
0.252 |
|
|
|
| 9 |
H |
0.252 |
|
|
|
| 10 |
H |
0.252 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-5.052 |
5.052 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.920 |
0.000 |
0.000 |
| y |
0.000 |
-37.078 |
0.000 |
| z |
0.000 |
0.000 |
-34.692 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.965 |
0.000 |
0.000 |
| y |
0.000 |
-3.772 |
0.000 |
| z |
0.000 |
0.000 |
-0.193 |
|
| Polar |
|---|
| 3z2-r2 | -0.386 |
|---|
| x2-y2 | 5.158 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.560 |
0.000 |
0.000 |
| y |
0.000 |
4.529 |
0.000 |
| z |
0.000 |
0.000 |
6.932 |
<r2> (average value of r
2) Å
2
| <r2> |
132.386 |
| (<r2>)1/2 |
11.506 |
Jump to
S1C1
Energy calculated at mPW1PW91/3-21G*
| | hartrees |
|---|
| Energy at 0K | -340.416046 |
| Energy at 298.15K | -340.422293 |
| HF Energy | -340.416046 |
| Nuclear repulsion energy | 242.000089 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3177 |
3017 |
8.05 |
|
|
|
| 2 |
A |
3096 |
2940 |
17.95 |
|
|
|
| 3 |
A |
1948 |
1850 |
459.30 |
|
|
|
| 4 |
A |
1579 |
1500 |
1.32 |
|
|
|
| 5 |
A |
1401 |
1330 |
4.58 |
|
|
|
| 6 |
A |
1273 |
1209 |
17.27 |
|
|
|
| 7 |
A |
1168 |
1109 |
27.73 |
|
|
|
| 8 |
A |
1123 |
1066 |
132.67 |
|
|
|
| 9 |
A |
937 |
889 |
2.94 |
|
|
|
| 10 |
A |
782 |
742 |
31.66 |
|
|
|
| 11 |
A |
706 |
670 |
2.39 |
|
|
|
| 12 |
A |
126 |
120 |
0.71 |
|
|
|
| 13 |
B |
3188 |
3027 |
20.34 |
|
|
|
| 14 |
B |
3100 |
2944 |
38.89 |
|
|
|
| 15 |
B |
1572 |
1492 |
3.97 |
|
|
|
| 16 |
B |
1415 |
1344 |
0.32 |
|
|
|
| 17 |
B |
1267 |
1203 |
1.05 |
|
|
|
| 18 |
B |
1128 |
1072 |
6.46 |
|
|
|
| 19 |
B |
971 |
922 |
210.85 |
|
|
|
| 20 |
B |
909 |
863 |
67.32 |
|
|
|
| 21 |
B |
741 |
704 |
27.98 |
|
|
|
| 22 |
B |
668 |
635 |
0.65 |
|
|
|
| 23 |
B |
489 |
464 |
0.57 |
|
|
|
| 24 |
B |
137 |
130 |
4.07 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16449.0 cm
-1
Scaled (by 0.9496) Zero Point Vibrational Energy (zpe) 15620.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at mPW1PW91/3-21G*
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.886 |
| O2 |
0.000 |
0.000 |
2.081 |
| O3 |
0.000 |
1.154 |
0.046 |
| O4 |
0.000 |
-1.154 |
0.046 |
| C5 |
0.216 |
0.743 |
-1.304 |
| C6 |
-0.216 |
-0.743 |
-1.304 |
| H7 |
-0.390 |
1.357 |
-1.968 |
| H8 |
1.275 |
0.844 |
-1.557 |
| H9 |
0.390 |
-1.357 |
-1.968 |
| H10 |
-1.275 |
-0.844 |
-1.557 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1949 | 1.4278 | 1.4278 | 2.3227 | 2.3227 | 3.1842 | 2.8821 | 3.1842 | 2.8821 |
O2 | 1.1949 | | 2.3397 | 2.3397 | 3.4722 | 3.4722 | 4.2881 | 3.9461 | 4.2881 | 3.9461 | O3 | 1.4278 | 2.3397 | | 2.3087 | 1.4278 | 2.3383 | 2.0614 | 2.0716 | 3.2424 | 2.8615 | O4 | 1.4278 | 2.3397 | 2.3087 | | 2.3383 | 1.4278 | 3.2424 | 2.8615 | 2.0614 | 2.0716 | C5 | 2.3227 | 3.4722 | 1.4278 | 2.3383 | | 1.5469 | 1.0886 | 1.0937 | 2.2086 | 2.1922 | C6 | 2.3227 | 3.4722 | 2.3383 | 1.4278 | 1.5469 | | 2.2086 | 2.1922 | 1.0886 | 1.0937 | H7 | 3.1842 | 4.2881 | 2.0614 | 3.2424 | 1.0886 | 2.2086 | | 1.7903 | 2.8228 | 2.4073 | H8 | 2.8821 | 3.9461 | 2.0716 | 2.8615 | 1.0937 | 2.1922 | 1.7903 | | 2.4073 | 3.0587 | H9 | 3.1842 | 4.2881 | 3.2424 | 2.0614 | 2.2086 | 1.0886 | 2.8228 | 2.4073 | | 1.7903 | H10 | 2.8821 | 3.9461 | 2.8615 | 2.0716 | 2.1922 | 1.0937 | 2.4073 | 3.0587 | 1.7903 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
108.861 |
|
C1 |
O4 |
C6 |
108.861 |
| O2 |
C1 |
O3 |
126.050 |
|
O2 |
C1 |
O4 |
126.050 |
| O3 |
C1 |
O4 |
107.900 |
|
O3 |
C5 |
C6 |
103.565 |
| O3 |
C5 |
H7 |
109.265 |
|
O3 |
C5 |
H8 |
109.779 |
| O4 |
C6 |
C5 |
103.565 |
|
O4 |
C6 |
H9 |
109.265 |
| O4 |
C6 |
H10 |
109.779 |
|
C5 |
C6 |
H9 |
112.698 |
| C5 |
C6 |
H10 |
111.070 |
|
C6 |
C5 |
H7 |
112.698 |
| C6 |
C5 |
H8 |
111.070 |
|
H7 |
C5 |
H8 |
110.242 |
| H9 |
C6 |
H10 |
110.242 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.887 |
|
|
|
| 2 |
O |
-0.472 |
|
|
|
| 3 |
O |
-0.504 |
|
|
|
| 4 |
O |
-0.504 |
|
|
|
| 5 |
C |
-0.209 |
|
|
|
| 6 |
C |
-0.209 |
|
|
|
| 7 |
H |
0.255 |
|
|
|
| 8 |
H |
0.250 |
|
|
|
| 9 |
H |
0.255 |
|
|
|
| 10 |
H |
0.250 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-4.976 |
4.976 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.960 |
0.265 |
0.000 |
| y |
0.265 |
-37.130 |
0.000 |
| z |
0.000 |
0.000 |
-34.505 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.857 |
0.265 |
0.000 |
| y |
0.265 |
-3.897 |
0.000 |
| z |
0.000 |
0.000 |
0.040 |
|
| Polar |
|---|
| 3z2-r2 | 0.081 |
|---|
| x2-y2 | 5.169 |
|---|
| xy | 0.265 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.648 |
0.128 |
0.000 |
| y |
0.128 |
4.458 |
0.000 |
| z |
0.000 |
0.000 |
6.901 |
<r2> (average value of r
2) Å
2
| <r2> |
131.117 |
| (<r2>)1/2 |
11.451 |