Jump to
S1C2
Energy calculated at mPW1PW91/6-31G
| | hartrees |
|---|
| Energy at 0K | -551.236803 |
| Energy at 298.15K | |
| HF Energy | -551.236803 |
| Nuclear repulsion energy | 348.775750 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/6-31G
Geometric Data calculated at mPW1PW91/6-31G
Point Group is D4h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
1.017 |
0.000 |
| C2 |
1.017 |
0.000 |
0.000 |
| C3 |
0.000 |
-1.017 |
0.000 |
| C4 |
-1.017 |
0.000 |
0.000 |
| F5 |
0.000 |
2.363 |
0.000 |
| F6 |
2.363 |
0.000 |
0.000 |
| F7 |
0.000 |
-2.363 |
0.000 |
| F8 |
-2.363 |
0.000 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
| C1 | | 1.4389 | 2.0350 | 1.4389 | 1.3453 | 2.5725 | 3.3802 | 2.5725 |
C2 | 1.4389 | | 1.4389 | 2.0350 | 2.5725 | 1.3453 | 2.5725 | 3.3802 | C3 | 2.0350 | 1.4389 | | 1.4389 | 3.3802 | 2.5725 | 1.3453 | 2.5725 | C4 | 1.4389 | 2.0350 | 1.4389 | | 2.5725 | 3.3802 | 2.5725 | 1.3453 | F5 | 1.3453 | 2.5725 | 3.3802 | 2.5725 | | 3.3415 | 4.7255 | 3.3415 | F6 | 2.5725 | 1.3453 | 2.5725 | 3.3802 | 3.3415 | | 3.3415 | 4.7255 | F7 | 3.3802 | 2.5725 | 1.3453 | 2.5725 | 4.7255 | 3.3415 | | 3.3415 | F8 | 2.5725 | 3.3802 | 2.5725 | 1.3453 | 3.3415 | 4.7255 | 3.3415 | |
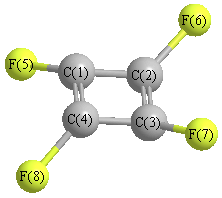 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
90.000 |
|
C1 |
C2 |
F6 |
135.000 |
| C1 |
C4 |
C3 |
90.000 |
|
C1 |
C4 |
F8 |
135.000 |
| C2 |
C1 |
C4 |
90.000 |
|
C2 |
C1 |
F5 |
135.000 |
| C2 |
C3 |
C4 |
90.000 |
|
C2 |
C3 |
F7 |
135.000 |
| C3 |
C2 |
F6 |
135.000 |
|
C3 |
C4 |
F8 |
135.000 |
| C4 |
C1 |
F5 |
135.000 |
|
C4 |
C3 |
F7 |
135.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.422 |
|
|
|
| 2 |
C |
0.106 |
|
|
|
| 3 |
C |
0.422 |
|
|
|
| 4 |
C |
0.106 |
|
|
|
| 5 |
F |
-0.229 |
|
|
|
| 6 |
F |
-0.299 |
|
|
|
| 7 |
F |
-0.229 |
|
|
|
| 8 |
F |
-0.299 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-50.589 |
0.000 |
0.000 |
| y |
0.000 |
-39.861 |
0.000 |
| z |
0.000 |
0.000 |
-40.453 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-10.432 |
0.000 |
0.000 |
| y |
0.000 |
5.660 |
0.000 |
| z |
0.000 |
0.000 |
4.772 |
|
| Polar |
|---|
| 3z2-r2 | 9.544 |
|---|
| x2-y2 | -10.728 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.823 |
0.000 |
0.000 |
| y |
0.000 |
7.735 |
0.000 |
| z |
0.000 |
0.000 |
2.522 |
<r2> (average value of r
2) Å
2
| <r2> |
253.075 |
| (<r2>)1/2 |
15.908 |
Jump to
S1C1
Energy calculated at mPW1PW91/6-31G
| | hartrees |
|---|
| Energy at 0K | -551.317773 |
| Energy at 298.15K | -551.317463 |
| HF Energy | -551.317773 |
| Nuclear repulsion energy | 347.992644 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at mPW1PW91/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
1909 |
1808 |
0.00 |
|
|
|
| 2 |
Ag |
1240 |
1174 |
0.00 |
|
|
|
| 3 |
Ag |
635 |
601 |
0.00 |
|
|
|
| 4 |
Ag |
231 |
218 |
0.00 |
|
|
|
| 5 |
Ag |
121 |
115 |
0.00 |
|
|
|
| 6 |
Au |
1314 |
1243 |
330.74 |
|
|
|
| 7 |
Au |
893 |
846 |
33.10 |
|
|
|
| 8 |
Au |
566 |
536 |
16.54 |
|
|
|
| 9 |
Au |
215 |
203 |
0.52 |
|
|
|
| 10 |
Au |
157 |
149 |
0.03 |
|
|
|
| 11 |
Bg |
1378 |
1304 |
0.00 |
|
|
|
| 12 |
Bg |
742 |
702 |
0.00 |
|
|
|
| 13 |
Bg |
483 |
458 |
0.00 |
|
|
|
| 14 |
Bg |
443 |
419 |
0.00 |
|
|
|
| 15 |
Bu |
1892 |
1791 |
74.22 |
|
|
|
| 16 |
Bu |
963 |
912 |
145.95 |
|
|
|
| 17 |
Bu |
291 |
276 |
8.75 |
|
|
|
| 18 |
Bu |
197 |
187 |
1.92 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 6835.1 cm
-1
Scaled (by 0.9466) Zero Point Vibrational Energy (zpe) 6470.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at mPW1PW91/6-31G
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.062 |
0.787 |
0.664 |
| C2 |
-0.062 |
-0.787 |
0.664 |
| C3 |
-0.062 |
-0.787 |
-0.664 |
| C4 |
0.062 |
0.787 |
-0.664 |
| F5 |
-0.062 |
1.685 |
1.659 |
| F6 |
0.062 |
-1.685 |
1.659 |
| F7 |
0.062 |
-1.685 |
-1.659 |
| F8 |
-0.062 |
1.685 |
-1.659 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
| C1 | | 1.5787 | 2.0624 | 1.3270 | 1.3467 | 2.6652 | 3.3921 | 2.4933 |
C2 | 1.5787 | | 1.3270 | 2.0624 | 2.6652 | 1.3467 | 2.4933 | 3.3921 | C3 | 2.0624 | 1.3270 | | 1.5787 | 3.3921 | 2.4933 | 1.3467 | 2.6652 | C4 | 1.3270 | 2.0624 | 1.5787 | | 2.4933 | 3.3921 | 2.6652 | 1.3467 | F5 | 1.3467 | 2.6652 | 3.3921 | 2.4933 | | 3.3730 | 4.7314 | 3.3180 | F6 | 2.6652 | 1.3467 | 2.4933 | 3.3921 | 3.3730 | | 3.3180 | 4.7314 | F7 | 3.3921 | 2.4933 | 1.3467 | 2.6652 | 4.7314 | 3.3180 | | 3.3730 | F8 | 2.4933 | 3.3921 | 2.6652 | 1.3467 | 3.3180 | 4.7314 | 3.3730 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
90.000 |
|
C1 |
C2 |
F6 |
131.130 |
| C1 |
C4 |
C3 |
90.000 |
|
C1 |
C4 |
F8 |
137.664 |
| C2 |
C1 |
C4 |
90.000 |
|
C2 |
C1 |
F5 |
131.130 |
| C2 |
C3 |
C4 |
90.000 |
|
C2 |
C3 |
F7 |
137.664 |
| C3 |
C2 |
F6 |
137.664 |
|
C3 |
C4 |
F8 |
131.130 |
| C4 |
C1 |
F5 |
137.664 |
|
C4 |
C3 |
F7 |
131.130 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at mPW1PW91/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.271 |
|
|
|
| 2 |
C |
0.271 |
|
|
|
| 3 |
C |
0.271 |
|
|
|
| 4 |
C |
0.271 |
|
|
|
| 5 |
F |
-0.271 |
|
|
|
| 6 |
F |
-0.271 |
|
|
|
| 7 |
F |
-0.271 |
|
|
|
| 8 |
F |
-0.271 |
|
|
|
Electric dipole moments
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.122 |
0.076 |
0.000 |
| y |
0.076 |
-45.222 |
0.000 |
| z |
0.000 |
0.000 |
-45.505 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.241 |
0.076 |
0.000 |
| y |
0.076 |
-2.409 |
0.000 |
| z |
0.000 |
0.000 |
-2.832 |
|
| Polar |
|---|
| 3z2-r2 | -5.664 |
|---|
| x2-y2 | 5.100 |
|---|
| xy | 0.076 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.452 |
-0.070 |
0.000 |
| y |
-0.070 |
5.344 |
0.000 |
| z |
0.000 |
0.000 |
7.418 |
<r2> (average value of r
2) Å
2
| <r2> |
254.239 |
| (<r2>)1/2 |
15.945 |