Jump to
S1C2
Energy calculated at MP2/CEP-31G
| | hartrees |
|---|
| Energy at 0K | -69.918651 |
| Energy at 298.15K | -69.923669 |
| HF Energy | -69.322709 |
| Nuclear repulsion energy | 120.910739 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/CEP-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3760 |
3633 |
66.00 |
|
|
|
| 2 |
A' |
3631 |
3508 |
67.21 |
|
|
|
| 3 |
A' |
3600 |
3478 |
78.79 |
|
|
|
| 4 |
A' |
1694 |
1637 |
191.95 |
|
|
|
| 5 |
A' |
1621 |
1567 |
97.58 |
|
|
|
| 6 |
A' |
1596 |
1543 |
202.96 |
|
|
|
| 7 |
A' |
1398 |
1351 |
2.66 |
|
|
|
| 8 |
A' |
1299 |
1255 |
77.87 |
|
|
|
| 9 |
A' |
1113 |
1076 |
141.31 |
|
|
|
| 10 |
A' |
1078 |
1042 |
201.00 |
|
|
|
| 11 |
A' |
742 |
717 |
0.44 |
|
|
|
| 12 |
A' |
572 |
553 |
67.67 |
|
|
|
| 13 |
A' |
514 |
497 |
1.27 |
|
|
|
| 14 |
A' |
399 |
386 |
8.03 |
|
|
|
| 15 |
A' |
256 |
247 |
11.81 |
|
|
|
| 16 |
A" |
760 |
735 |
0.56 |
|
|
|
| 17 |
A" |
637 |
615 |
356.50 |
|
|
|
| 18 |
A" |
601 |
581 |
6.94 |
|
|
|
| 19 |
A" |
578 |
558 |
257.03 |
|
|
|
| 20 |
A" |
401 |
388 |
8.84 |
|
|
|
| 21 |
A" |
61 |
59 |
7.24 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13155.4 cm
-1
Scaled (by 0.9663) Zero Point Vibrational Energy (zpe) 12712.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/CEP-31G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.765 |
0.000 |
| C2 |
-0.047 |
-0.800 |
0.000 |
| O3 |
-1.131 |
-1.479 |
0.000 |
| O4 |
1.081 |
1.441 |
0.000 |
| O5 |
-1.275 |
1.337 |
0.000 |
| N6 |
1.241 |
-1.344 |
0.000 |
| H7 |
1.341 |
-2.358 |
0.000 |
| H8 |
2.065 |
-0.743 |
0.000 |
| H9 |
-1.213 |
2.328 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5658 | 2.5127 | 1.2755 | 1.3972 | 2.4468 | 3.3986 | 2.5571 | 1.9789 |
C2 | 1.5658 | | 1.2794 | 2.5096 | 2.4650 | 1.3978 | 2.0862 | 2.1129 | 3.3390 | O3 | 2.5127 | 1.2794 | | 3.6639 | 2.8195 | 2.3763 | 2.6239 | 3.2804 | 3.8080 | O4 | 1.2755 | 2.5096 | 3.6639 | | 2.3581 | 2.7900 | 3.8086 | 2.3959 | 2.4596 | O5 | 1.3972 | 2.4650 | 2.8195 | 2.3581 | | 3.6764 | 4.5272 | 3.9347 | 0.9932 | N6 | 2.4468 | 1.3978 | 2.3763 | 2.7900 | 3.6764 | | 1.0192 | 1.0201 | 4.4167 | H7 | 3.3986 | 2.0862 | 2.6239 | 3.8086 | 4.5272 | 1.0192 | | 1.7704 | 5.3372 | H8 | 2.5571 | 2.1129 | 3.2804 | 2.3959 | 3.9347 | 1.0201 | 1.7704 | | 4.4922 | H9 | 1.9789 | 3.3390 | 3.8080 | 2.4596 | 0.9932 | 4.4167 | 5.3372 | 4.4922 | |
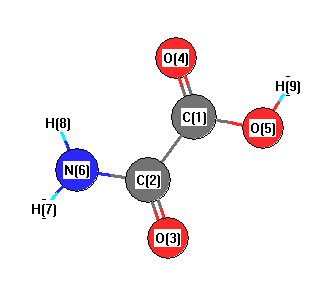 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
123.735 |
|
C1 |
C2 |
N6 |
111.172 |
| C1 |
O5 |
H9 |
110.619 |
|
C2 |
C1 |
O4 |
123.754 |
| C2 |
C1 |
O5 |
112.470 |
|
C2 |
N6 |
H7 |
118.495 |
| C2 |
N6 |
H8 |
121.025 |
|
O3 |
C2 |
N6 |
125.092 |
| O4 |
C1 |
O5 |
123.776 |
|
H7 |
N6 |
H8 |
120.479 |
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2/CEP-31G
| | hartrees |
|---|
| Energy at 0K | -69.921357 |
| Energy at 298.15K | -69.926505 |
| Nuclear repulsion energy | 121.558025 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/CEP-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3751 |
3625 |
73.16 |
|
|
|
| 2 |
A' |
3592 |
3471 |
96.86 |
|
|
|
| 3 |
A' |
3571 |
3451 |
91.72 |
|
|
|
| 4 |
A' |
1700 |
1643 |
233.62 |
|
|
|
| 5 |
A' |
1651 |
1595 |
194.18 |
|
|
|
| 6 |
A' |
1614 |
1560 |
1.14 |
|
|
|
| 7 |
A' |
1396 |
1349 |
17.61 |
|
|
|
| 8 |
A' |
1252 |
1210 |
518.11 |
|
|
|
| 9 |
A' |
1138 |
1100 |
40.30 |
|
|
|
| 10 |
A' |
1094 |
1057 |
10.11 |
|
|
|
| 11 |
A' |
751 |
726 |
21.63 |
|
|
|
| 12 |
A' |
585 |
566 |
10.49 |
|
|
|
| 13 |
A' |
523 |
505 |
2.13 |
|
|
|
| 14 |
A' |
389 |
376 |
4.75 |
|
|
|
| 15 |
A' |
254 |
246 |
38.45 |
|
|
|
| 16 |
A" |
747 |
722 |
1.48 |
|
|
|
| 17 |
A" |
653 |
631 |
334.35 |
|
|
|
| 18 |
A" |
626 |
605 |
256.72 |
|
|
|
| 19 |
A" |
615 |
594 |
18.71 |
|
|
|
| 20 |
A" |
405 |
392 |
8.02 |
|
|
|
| 21 |
A" |
103 |
100 |
10.40 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13205.9 cm
-1
Scaled (by 0.9663) Zero Point Vibrational Energy (zpe) 12760.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/CEP-31G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.019 |
-0.804 |
0.000 |
| C2 |
0.000 |
0.771 |
0.000 |
| O3 |
-1.123 |
1.402 |
0.000 |
| O4 |
1.080 |
-1.495 |
0.000 |
| O5 |
-1.260 |
-1.365 |
0.000 |
| N6 |
1.261 |
1.343 |
0.000 |
| H7 |
1.348 |
2.359 |
0.000 |
| H8 |
2.092 |
0.751 |
0.000 |
| H9 |
-1.956 |
-0.652 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5743 | 2.4841 | 1.2663 | 1.3977 | 2.4796 | 3.4304 | 2.5911 | 1.9813 |
C2 | 1.5743 | | 1.2885 | 2.5100 | 2.4800 | 1.3845 | 2.0834 | 2.0924 | 2.4190 | O3 | 2.4841 | 1.2885 | | 3.6399 | 2.7708 | 2.3843 | 2.6500 | 3.2806 | 2.2171 | O4 | 1.2663 | 2.5100 | 3.6399 | | 2.3444 | 2.8438 | 3.8633 | 2.4635 | 3.1511 | O5 | 1.3977 | 2.4800 | 2.7708 | 2.3444 | | 3.7000 | 4.5470 | 3.9648 | 0.9959 | N6 | 2.4796 | 1.3845 | 2.3843 | 2.8438 | 3.7000 | | 1.0197 | 1.0210 | 3.7854 | H7 | 3.4304 | 2.0834 | 2.6500 | 3.8633 | 4.5470 | 1.0197 | | 1.7717 | 4.4707 | H8 | 2.5911 | 2.0924 | 3.2806 | 2.4635 | 3.9648 | 1.0210 | 1.7717 | | 4.2848 | H9 | 1.9813 | 2.4190 | 2.2171 | 3.1511 | 0.9959 | 3.7854 | 4.4707 | 4.2848 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
120.062 |
|
C1 |
C2 |
N6 |
113.718 |
| C1 |
O5 |
H9 |
110.611 |
|
C2 |
C1 |
O4 |
123.803 |
| C2 |
C1 |
O5 |
112.985 |
|
C2 |
N6 |
H7 |
119.361 |
| C2 |
N6 |
H8 |
120.132 |
|
O3 |
C2 |
N6 |
126.220 |
| O4 |
C1 |
O5 |
123.211 |
|
H7 |
N6 |
H8 |
120.506 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability