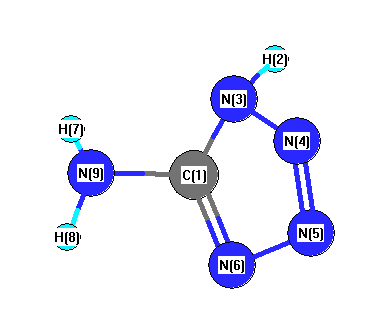
Vibrational Frequencies calculated at MP2/CEP-121G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3648 |
3476 |
93.42 |
|
|
|
| 2 |
A |
3638 |
3466 |
28.64 |
|
|
|
| 3 |
A |
3533 |
3366 |
21.67 |
|
|
|
| 4 |
A |
1711 |
1631 |
84.28 |
|
|
|
| 5 |
A |
1602 |
1526 |
147.03 |
|
|
|
| 6 |
A |
1501 |
1430 |
29.79 |
|
|
|
| 7 |
A |
1363 |
1299 |
0.44 |
|
|
|
| 8 |
A |
1186 |
1130 |
3.30 |
|
|
|
| 9 |
A |
1155 |
1101 |
24.60 |
|
|
|
| 10 |
A |
1130 |
1077 |
7.28 |
|
|
|
| 11 |
A |
1093 |
1041 |
16.94 |
|
|
|
| 12 |
A |
1056 |
1006 |
16.27 |
|
|
|
| 13 |
A |
988 |
941 |
0.39 |
|
|
|
| 14 |
A |
869 |
828 |
232.66 |
|
|
|
| 15 |
A |
716 |
682 |
7.10 |
|
|
|
| 16 |
A |
700 |
667 |
9.08 |
|
|
|
| 17 |
A |
683 |
651 |
59.78 |
|
|
|
| 18 |
A |
561 |
534 |
67.87 |
|
|
|
| 19 |
A |
378 |
361 |
16.47 |
|
|
|
| 20 |
A |
288 |
274 |
1.86 |
|
|
|
| 21 |
A |
197 |
188 |
61.92 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13998.0 cm
-1
Scaled (by 0.9528) Zero Point Vibrational Energy (zpe) 13337.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.