Vibrational Frequencies calculated at MP2/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3279 |
3130 |
4.65 |
|
|
|
| 2 |
A |
3245 |
3097 |
0.42 |
|
|
|
| 3 |
A |
3221 |
3075 |
14.12 |
|
|
|
| 4 |
A |
3182 |
3037 |
2.42 |
|
|
|
| 5 |
A |
3177 |
3033 |
22.98 |
|
|
|
| 6 |
A |
3105 |
2964 |
19.01 |
|
|
|
| 7 |
A |
1713 |
1635 |
72.79 |
|
|
|
| 8 |
A |
1683 |
1607 |
12.88 |
|
|
|
| 9 |
A |
1601 |
1528 |
7.45 |
|
|
|
| 10 |
A |
1587 |
1514 |
5.88 |
|
|
|
| 11 |
A |
1525 |
1455 |
10.83 |
|
|
|
| 12 |
A |
1506 |
1437 |
34.59 |
|
|
|
| 13 |
A |
1361 |
1299 |
23.89 |
|
|
|
| 14 |
A |
1207 |
1152 |
450.72 |
|
|
|
| 15 |
A |
1204 |
1149 |
3.20 |
|
|
|
| 16 |
A |
1190 |
1136 |
0.33 |
|
|
|
| 17 |
A |
1122 |
1071 |
3.37 |
|
|
|
| 18 |
A |
1045 |
998 |
38.62 |
|
|
|
| 19 |
A |
1006 |
960 |
23.75 |
|
|
|
| 20 |
A |
987 |
942 |
11.43 |
|
|
|
| 21 |
A |
827 |
790 |
10.27 |
|
|
|
| 22 |
A |
816 |
779 |
31.14 |
|
|
|
| 23 |
A |
667 |
637 |
5.16 |
|
|
|
| 24 |
A |
531 |
507 |
0.11 |
|
|
|
| 25 |
A |
481 |
459 |
0.64 |
|
|
|
| 26 |
A |
330 |
315 |
21.11 |
|
|
|
| 27 |
A |
213 |
204 |
3.12 |
|
|
|
| 28 |
A |
199 |
190 |
7.00 |
|
|
|
| 29 |
A |
107 |
102 |
3.82 |
|
|
|
| 30 |
A |
85 |
81 |
0.06 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21101.1 cm
-1
Scaled (by 0.9545) Zero Point Vibrational Energy (zpe) 20141.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
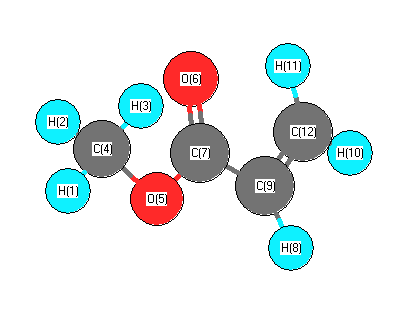
Charges from optimized geometry at MP2/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.203 |
|
|
0.109 |
| 2 |
H |
0.201 |
|
|
0.082 |
| 3 |
H |
0.201 |
|
|
0.082 |
| 4 |
C |
-0.303 |
|
|
-0.040 |
| 5 |
O |
-0.530 |
|
|
-0.469 |
| 6 |
O |
-0.469 |
|
|
-0.493 |
| 7 |
C |
0.652 |
|
|
0.892 |
| 8 |
H |
0.223 |
|
|
0.204 |
| 9 |
C |
-0.266 |
|
|
-0.447 |
| 10 |
H |
0.198 |
|
|
0.164 |
| 11 |
H |
0.214 |
|
|
0.141 |
| 12 |
C |
-0.324 |
|
|
-0.226 |
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.305 |
-0.994 |
0.000 |
1.040 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
-0.325 |
-0.975 |
0.000 |
1.028 |
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-26.729 |
-1.038 |
-0.002 |
| y |
-1.038 |
-39.087 |
-0.009 |
| z |
-0.002 |
-0.009 |
-36.719 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
11.174 |
-1.038 |
-0.002 |
| y |
-1.038 |
-7.363 |
-0.009 |
| z |
-0.002 |
-0.009 |
-3.811 |
|
| Polar |
|---|
| 3z2-r2 | -7.622 |
|---|
| x2-y2 | 12.358 |
|---|
| xy | -1.038 |
|---|
| xz | -0.002 |
|---|
| yz | -0.009 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.421 |
0.536 |
-0.002 |
| y |
0.536 |
6.247 |
-0.003 |
| z |
-0.002 |
-0.003 |
2.997 |
<r2> (average value of r
2) Å
2
| <r2> |
181.637 |
| (<r2>)1/2 |
13.477 |