Jump to
S1C2
Energy calculated at MP2/STO-3G
| | hartrees |
|---|
| Energy at 0K | -738.102640 |
| Energy at 298.15K | -738.102840 |
| HF Energy | -737.758978 |
| Nuclear repulsion energy | 545.656447 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
1816 |
1584 |
0.00 |
|
|
|
| 2 |
Ag |
1551 |
1352 |
0.00 |
|
|
|
| 3 |
Ag |
1445 |
1260 |
0.00 |
|
|
|
| 4 |
Ag |
1246 |
1086 |
0.00 |
|
|
|
| 5 |
Ag |
717 |
625 |
0.00 |
|
|
|
| 6 |
Ag |
557 |
485 |
0.00 |
|
|
|
| 7 |
Ag |
373 |
325 |
0.00 |
|
|
|
| 8 |
Ag |
322 |
281 |
0.00 |
|
|
|
| 9 |
Ag |
174 |
152 |
0.00 |
|
|
|
| 10 |
Au |
492 |
429 |
4.31 |
|
|
|
| 11 |
Au |
323 |
282 |
2.41 |
|
|
|
| 12 |
Au |
112 |
98 |
0.01 |
|
|
|
| 13 |
Au |
30 |
26 |
0.02 |
|
|
|
| 14 |
Bg |
556 |
485 |
0.00 |
|
|
|
| 15 |
Bg |
448 |
390 |
0.00 |
|
|
|
| 16 |
Bg |
181 |
158 |
0.00 |
|
|
|
| 17 |
Bu |
1773 |
1546 |
116.82 |
|
|
|
| 18 |
Bu |
1502 |
1309 |
72.53 |
|
|
|
| 19 |
Bu |
1341 |
1169 |
91.66 |
|
|
|
| 20 |
Bu |
981 |
855 |
67.01 |
|
|
|
| 21 |
Bu |
606 |
528 |
7.45 |
|
|
|
| 22 |
Bu |
457 |
398 |
3.50 |
|
|
|
| 23 |
Bu |
258 |
225 |
2.31 |
|
|
|
| 24 |
Bu |
103 |
89 |
0.33 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8681.2 cm
-1
Scaled (by 0.8719) Zero Point Vibrational Energy (zpe) 7569.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/STO-3G
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.185 |
1.886 |
0.000 |
| C2 |
0.522 |
0.548 |
0.000 |
| C3 |
-0.522 |
-0.548 |
0.000 |
| C4 |
-0.185 |
-1.886 |
0.000 |
| F5 |
1.104 |
2.899 |
0.000 |
| F6 |
-1.104 |
2.342 |
0.000 |
| F7 |
1.847 |
0.161 |
0.000 |
| F8 |
-1.847 |
-0.161 |
0.000 |
| F9 |
1.104 |
-2.342 |
0.000 |
| F10 |
-1.104 |
-2.899 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
| C1 | | 1.3800 | 2.5342 | 3.7899 | 1.3685 | 1.3671 | 2.3957 | 2.8838 | 4.3265 | 4.9560 |
C2 | 1.3800 | | 1.5132 | 2.5342 | 2.4227 | 2.4213 | 1.3804 | 2.4725 | 2.9476 | 3.8115 | C3 | 2.5342 | 1.5132 | | 1.3800 | 3.8115 | 2.9476 | 2.4725 | 1.3804 | 2.4213 | 2.4227 | C4 | 3.7899 | 2.5342 | 1.3800 | | 4.9560 | 4.3265 | 2.8838 | 2.3957 | 1.3671 | 1.3685 | F5 | 1.3685 | 2.4227 | 3.8115 | 4.9560 | | 2.2777 | 2.8377 | 4.2513 | 5.2412 | 6.2052 | F6 | 1.3671 | 2.4213 | 2.9476 | 4.3265 | 2.2777 | | 3.6695 | 2.6104 | 5.1780 | 5.2412 | F7 | 2.3957 | 1.3804 | 2.4725 | 2.8838 | 2.8377 | 3.6695 | | 3.7077 | 2.6104 | 4.2513 | F8 | 2.8838 | 2.4725 | 1.3804 | 2.3957 | 4.2513 | 2.6104 | 3.7077 | | 3.6695 | 2.8377 | F9 | 4.3265 | 2.9476 | 2.4213 | 1.3671 | 5.2412 | 5.1780 | 2.6104 | 3.6695 | | 2.2777 | F10 | 4.9560 | 3.8115 | 2.4227 | 1.3685 | 6.2052 | 5.2412 | 4.2513 | 2.8377 | 2.2777 | |
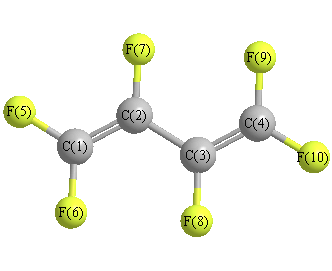 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
122.242 |
|
C1 |
C2 |
F7 |
120.427 |
| C2 |
C1 |
F5 |
123.643 |
|
C2 |
C1 |
F6 |
123.620 |
| C2 |
C3 |
C4 |
122.242 |
|
C2 |
C3 |
F8 |
117.331 |
| C3 |
C2 |
F7 |
117.331 |
|
C3 |
C4 |
F9 |
123.620 |
| C3 |
C4 |
F10 |
123.643 |
|
C4 |
C3 |
F8 |
120.427 |
| F5 |
C1 |
F6 |
112.737 |
|
F9 |
C4 |
F10 |
112.737 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2/STO-3G
| | hartrees |
|---|
| Energy at 0K | -738.100519 |
| Energy at 298.15K | -738.100787 |
| HF Energy | -737.756783 |
| Nuclear repulsion energy | 551.251653 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
1793 |
1564 |
26.26 |
|
|
|
| 2 |
A |
1524 |
1328 |
0.95 |
|
|
|
| 3 |
A |
1447 |
1262 |
70.10 |
|
|
|
| 4 |
A |
1202 |
1048 |
49.84 |
|
|
|
| 5 |
A |
706 |
615 |
0.14 |
|
|
|
| 6 |
A |
579 |
505 |
0.99 |
|
|
|
| 7 |
A |
478 |
417 |
0.38 |
|
|
|
| 8 |
A |
440 |
384 |
0.23 |
|
|
|
| 9 |
A |
364 |
318 |
1.82 |
|
|
|
| 10 |
A |
236 |
206 |
0.05 |
|
|
|
| 11 |
A |
193 |
168 |
0.09 |
|
|
|
| 12 |
A |
109 |
95 |
0.08 |
|
|
|
| 13 |
A |
44 |
38 |
0.02 |
|
|
|
| 14 |
B |
1795 |
1565 |
56.40 |
|
|
|
| 15 |
B |
1511 |
1318 |
62.55 |
|
|
|
| 16 |
B |
1365 |
1190 |
17.60 |
|
|
|
| 17 |
B |
1008 |
879 |
43.63 |
|
|
|
| 18 |
B |
609 |
531 |
4.96 |
|
|
|
| 19 |
B |
559 |
488 |
1.51 |
|
|
|
| 20 |
B |
503 |
438 |
4.76 |
|
|
|
| 21 |
B |
368 |
321 |
1.98 |
|
|
|
| 22 |
B |
286 |
249 |
1.01 |
|
|
|
| 23 |
B |
183 |
160 |
2.01 |
|
|
|
| 24 |
B |
111 |
97 |
0.18 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8706.5 cm
-1
Scaled (by 0.8719) Zero Point Vibrational Energy (zpe) 7591.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/STO-3G
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.072 |
1.558 |
-0.415 |
| C2 |
0.204 |
0.731 |
0.680 |
| C3 |
-0.204 |
-0.731 |
0.680 |
| C4 |
-0.072 |
-1.558 |
-0.415 |
| F5 |
0.467 |
2.868 |
-0.427 |
| F6 |
-0.467 |
1.162 |
-1.607 |
| F7 |
0.727 |
1.231 |
1.858 |
| F8 |
-0.727 |
-1.231 |
1.858 |
| F9 |
0.467 |
-1.162 |
-1.607 |
| F10 |
-0.467 |
-2.868 |
-0.427 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
| C1 | | 1.3793 | 2.5531 | 3.1203 | 1.3680 | 1.3668 | 2.3884 | 3.6861 | 2.9967 | 4.4591 |
C2 | 1.3793 | | 1.5177 | 2.5531 | 2.4212 | 2.4225 | 1.3823 | 2.4703 | 2.9811 | 3.8248 | C3 | 2.5531 | 1.5177 | | 1.3793 | 3.8248 | 2.9811 | 2.4703 | 1.3823 | 2.4225 | 2.4212 | C4 | 3.1203 | 2.5531 | 1.3793 | | 4.4591 | 2.9967 | 3.6861 | 2.3884 | 1.3668 | 1.3680 | F5 | 1.3680 | 2.4212 | 3.8248 | 4.4591 | | 2.2749 | 2.8229 | 4.8425 | 4.1994 | 5.8115 | F6 | 1.3668 | 2.4225 | 2.9811 | 2.9967 | 2.2749 | | 3.6658 | 4.2194 | 2.5057 | 4.1994 | F7 | 2.3884 | 1.3823 | 2.4703 | 3.6861 | 2.8229 | 3.6658 | | 2.8591 | 4.2194 | 4.8425 | F8 | 3.6861 | 2.4703 | 1.3823 | 2.3884 | 4.8425 | 4.2194 | 2.8591 | | 3.6658 | 2.8229 | F9 | 2.9967 | 2.9811 | 2.4225 | 1.3668 | 4.1994 | 2.5057 | 4.2194 | 3.6658 | | 2.2749 | F10 | 4.4591 | 3.8248 | 2.4212 | 1.3680 | 5.8115 | 4.1994 | 4.8425 | 2.8229 | 2.2749 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
123.523 |
|
C1 |
C2 |
F7 |
119.733 |
| C2 |
C1 |
F5 |
123.606 |
|
C2 |
C1 |
F6 |
123.813 |
| C2 |
C3 |
C4 |
123.523 |
|
C2 |
C3 |
F8 |
116.743 |
| C3 |
C2 |
F7 |
116.743 |
|
C3 |
C4 |
F9 |
123.813 |
| C3 |
C4 |
F10 |
123.606 |
|
C4 |
C3 |
F8 |
119.733 |
| F5 |
C1 |
F6 |
112.581 |
|
F9 |
C4 |
F10 |
112.581 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability