Jump to
S1C2
S1C3
Energy calculated at MP2/6-311G**
| | hartrees |
|---|
| Energy at 0K | -100.099080 |
| Energy at 298.15K | -100.098102 |
| HF Energy | -99.795235 |
| Nuclear repulsion energy | 27.286591 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP2/6-311G**
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
-2.082 |
| C2 |
0.000 |
0.000 |
-0.157 |
| N3 |
0.000 |
0.000 |
1.027 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 1.9248 | 3.1093 |
C2 | 1.9248 | | 1.1845 | N3 | 3.1093 | 1.1845 | |
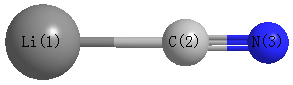 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
180.000 |
|
Li1 |
N3 |
C2 |
0.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at MP2/6-311G**
| | hartrees |
|---|
| Energy at 0K | -100.102924 |
| Energy at 298.15K | -100.102224 |
| HF Energy | -99.801985 |
| Nuclear repulsion energy | 29.048891 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP2/6-311G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
1.430 |
-0.596 |
0.000 |
| C2 |
-0.715 |
-0.376 |
0.000 |
| N3 |
0.000 |
0.577 |
0.000 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.1566 | 1.8497 |
C2 | 2.1566 | | 1.1913 | N3 | 1.8497 | 1.1913 | |
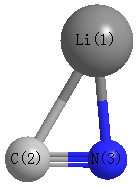 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
58.969 |
|
Li1 |
N3 |
C2 |
87.534 |
| C2 |
Li1 |
N3 |
33.496 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at MP2/6-311G**
| | hartrees |
|---|
| Energy at 0K | -100.099508 |
| Energy at 298.15K | -100.098269 |
| HF Energy | -99.807135 |
| Nuclear repulsion energy | 28.089418 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at MP2/6-311G**
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Li1 |
0.000 |
0.000 |
1.899 |
| C2 |
0.000 |
0.000 |
-1.079 |
| N3 |
0.000 |
0.000 |
0.111 |
Atom - Atom Distances (Å)
| |
Li1 |
C2 |
N3 |
| Li1 | | 2.9778 | 1.7876 |
C2 | 2.9778 | | 1.1902 | N3 | 1.7876 | 1.1902 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Li1 |
C2 |
N3 |
0.000 |
|
Li1 |
N3 |
C2 |
180.000 |
| C2 |
Li1 |
N3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability