Jump to
S1C2
Energy calculated at MP2/cc-pVQZ
| | hartrees |
|---|
| Energy at 0K | -275.186463 |
| Energy at 298.15K | |
| HF Energy | -274.256440 |
| Nuclear repulsion energy | 117.361416 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/cc-pVQZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2069 |
1962 |
44.50 |
72.09 |
0.10 |
0.18 |
| 2 |
A1 |
754 |
715 |
12.42 |
13.60 |
0.08 |
0.15 |
| 3 |
A1 |
534 |
506 |
73.65 |
0.90 |
0.21 |
0.34 |
| 4 |
A1 |
97 |
92 |
11.83 |
1.60 |
0.61 |
0.76 |
| 5 |
A2 |
464 |
440 |
0.00 |
2.61 |
0.75 |
0.86 |
| 6 |
B1 |
486 |
461 |
100.37 |
0.20 |
0.75 |
0.86 |
| 7 |
B2 |
2078 |
1971 |
1063.58 |
3.53 |
0.75 |
0.86 |
| 8 |
B2 |
1214 |
1152 |
105.74 |
0.84 |
0.75 |
0.86 |
| 9 |
B2 |
456 |
432 |
14.02 |
1.66 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 4075.6 cm
-1
Scaled (by 0.9484) Zero Point Vibrational Energy (zpe) 3865.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
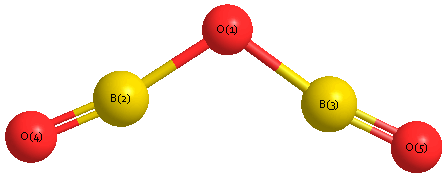
Geometric Data calculated at MP2/cc-pVQZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.570 |
| B2 |
0.000 |
1.236 |
0.075 |
| B3 |
0.000 |
-1.236 |
0.075 |
| O4 |
0.000 |
2.380 |
-0.332 |
| O5 |
0.000 |
-2.380 |
-0.332 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3312 | 1.3312 | 2.5455 | 2.5455 |
B2 | 1.3312 | | 2.4711 | 1.2148 | 3.6387 | B3 | 1.3312 | 2.4711 | | 3.6387 | 1.2148 | O4 | 2.5455 | 1.2148 | 3.6387 | | 4.7607 | O5 | 2.5455 | 3.6387 | 1.2148 | 4.7607 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
177.702 |
|
O1 |
B3 |
O5 |
177.702 |
| B2 |
O1 |
B3 |
136.300 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2/cc-pVQZ
| | hartrees |
|---|
| Energy at 0K | -275.183998 |
| Energy at 298.15K | |
| HF Energy | -274.255562 |
| Nuclear repulsion energy | 116.583154 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/cc-pVQZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2074 |
1967 |
0.00 |
97.95 |
0.12 |
0.22 |
| 2 |
Σg |
668 |
634 |
0.00 |
13.54 |
0.12 |
0.21 |
| 3 |
Σu |
2118 |
2009 |
1538.87 |
0.00 |
0.00 |
0.00 |
| 4 |
Σu |
1304 |
1237 |
106.44 |
0.00 |
0.00 |
0.00 |
| 5 |
Πg |
468 |
444 |
0.00 |
2.98 |
0.75 |
0.86 |
| 5 |
Πg |
468 |
444 |
0.00 |
2.98 |
0.75 |
0.86 |
| 6 |
Πu |
446 |
423 |
110.55 |
0.00 |
0.75 |
0.86 |
| 6 |
Πu |
446 |
423 |
110.55 |
0.00 |
0.75 |
0.86 |
| 7 |
Πu |
99i |
94i |
4.19 |
0.00 |
0.00 |
0.00 |
| 7 |
Πu |
99i |
94i |
4.19 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 3898.4 cm
-1
Scaled (by 0.9484) Zero Point Vibrational Energy (zpe) 3697.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/cc-pVQZ
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.000 |
| B2 |
0.000 |
0.000 |
1.313 |
| B3 |
0.000 |
0.000 |
-1.313 |
| O4 |
0.000 |
0.000 |
2.529 |
| O5 |
0.000 |
0.000 |
-2.529 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3131 | 1.3131 | 2.5292 | 2.5292 |
B2 | 1.3131 | | 2.6262 | 1.2161 | 3.8423 | B3 | 1.3131 | 2.6262 | | 3.8423 | 1.2161 | O4 | 2.5292 | 1.2161 | 3.8423 | | 5.0584 | O5 | 2.5292 | 3.8423 | 1.2161 | 5.0584 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
180.000 |
|
O1 |
B3 |
O5 |
180.000 |
| B2 |
O1 |
B3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability