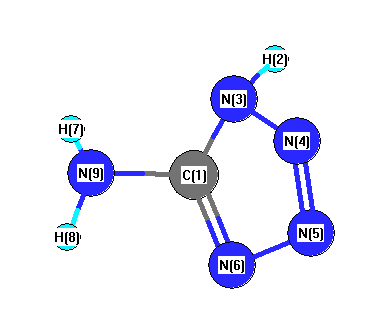
Vibrational Frequencies calculated at MP2/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3685 |
3493 |
39.45 |
|
|
|
| 2 |
A |
3679 |
3487 |
105.19 |
|
|
|
| 3 |
A |
3580 |
3394 |
33.59 |
|
|
|
| 4 |
A |
1672 |
1585 |
161.56 |
|
|
|
| 5 |
A |
1619 |
1535 |
92.27 |
|
|
|
| 6 |
A |
1509 |
1430 |
30.39 |
|
|
|
| 7 |
A |
1379 |
1307 |
0.58 |
|
|
|
| 8 |
A |
1194 |
1131 |
6.34 |
|
|
|
| 9 |
A |
1149 |
1089 |
25.09 |
|
|
|
| 10 |
A |
1122 |
1064 |
4.94 |
|
|
|
| 11 |
A |
1097 |
1040 |
11.69 |
|
|
|
| 12 |
A |
1066 |
1010 |
24.15 |
|
|
|
| 13 |
A |
1007 |
954 |
0.51 |
|
|
|
| 14 |
A |
804 |
762 |
206.27 |
|
|
|
| 15 |
A |
728 |
690 |
5.06 |
|
|
|
| 16 |
A |
697 |
661 |
13.41 |
|
|
|
| 17 |
A |
672 |
637 |
77.75 |
|
|
|
| 18 |
A |
454 |
431 |
53.60 |
|
|
|
| 19 |
A |
382 |
362 |
16.26 |
|
|
|
| 20 |
A |
285 |
270 |
2.27 |
|
|
|
| 21 |
A |
219 |
207 |
50.25 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13998.2 cm
-1
Scaled (by 0.9479) Zero Point Vibrational Energy (zpe) 13268.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.