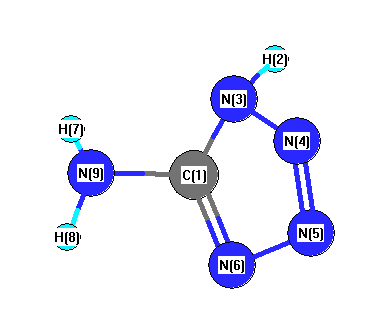
Vibrational Frequencies calculated at MP2/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3682 |
3503 |
46.18 |
|
|
|
| 2 |
A |
3677 |
3499 |
101.90 |
|
|
|
| 3 |
A |
3578 |
3404 |
38.07 |
|
|
|
| 4 |
A |
1666 |
1585 |
172.07 |
|
|
|
| 5 |
A |
1616 |
1538 |
77.19 |
|
|
|
| 6 |
A |
1510 |
1437 |
25.23 |
|
|
|
| 7 |
A |
1367 |
1301 |
1.10 |
|
|
|
| 8 |
A |
1193 |
1136 |
6.52 |
|
|
|
| 9 |
A |
1159 |
1102 |
18.05 |
|
|
|
| 10 |
A |
1117 |
1063 |
6.08 |
|
|
|
| 11 |
A |
1101 |
1048 |
11.68 |
|
|
|
| 12 |
A |
1066 |
1014 |
22.80 |
|
|
|
| 13 |
A |
1010 |
961 |
0.21 |
|
|
|
| 14 |
A |
780 |
742 |
110.25 |
|
|
|
| 15 |
A |
731 |
696 |
3.61 |
|
|
|
| 16 |
A |
727 |
692 |
3.57 |
|
|
|
| 17 |
A |
683 |
650 |
158.01 |
|
|
|
| 18 |
A |
570 |
543 |
59.07 |
|
|
|
| 19 |
A |
386 |
367 |
10.36 |
|
|
|
| 20 |
A |
307 |
292 |
1.94 |
|
|
|
| 21 |
A |
225 |
214 |
48.23 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14076.2 cm
-1
Scaled (by 0.9515) Zero Point Vibrational Energy (zpe) 13393.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.